








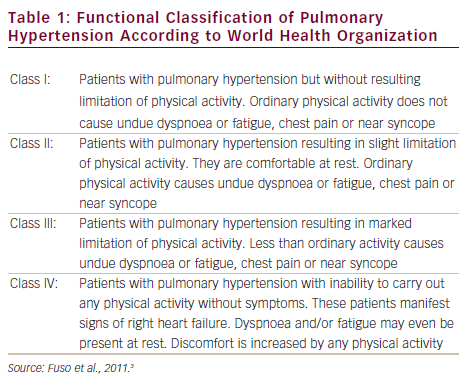
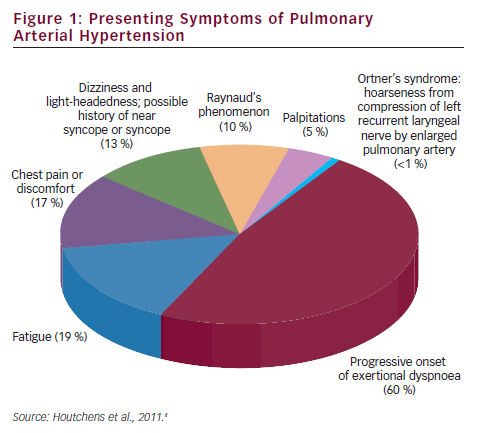
Pulmonary arterial hypertension (PAH) is a clinical syndrome characterised by a progressive increase of pulmonary vascular resistance, ultimately leading to right heart failure and death if left untreated.1 According to the guidelines published by the European Society of Cardiology (ESC) and the European Respiratory Society (ERS),2 PAH is characterised by pre-capillary pulmonary hypertension (mean pulmonary arterial pressure [PAP] ≥25 mmHg, with pulmonary wedge pressure ≤15 mmHg) in the absence of left heart disease, parenchymal lung diseases and thromboembolic disease. PAH includes idiopathic forms, heriditable forms, or can be associated with systemic diseases such as connective tissue diseases, HIV infection, portal hypertension, congenital heart diseases, schistosomiasis and haemolytic anemias.3 The symptoms of PAH are non-specific and include breathlessness, fatigue, weakness, angina, syncope and abdominal distension (see Figure 1).4,5 The severity of the disease can be classified by a functional class (FC) defined by the World Health Organization (WHO) (see Table 1).3 Symptoms are usually delayed in appearance and progress slowly, with the result that diagnosis of PAH typically occurs at advanced stages: FC III or FC IV.
Endothelial and platelet dysfunction play a key role in PAH pathogenesis, inducing a sustained impaired production of vasodilator and antiproliferative agents such as nitric oxide and prostacyclin, along with overexpression of vasoconstrictor and promitotic molecules such as endothelin.6 According to these pathogenetic pathways, three classes of drugs have been developed: endothelin receptor antagonists (ERA), phosphodiesterase-5 (PDE5) inhibitors and prostanoids. Although none of these therapies are curative, several randomised controlled trials (RCTs) have demonstrated their clinical efficacy for the treatment of this deadly disease.
Given the availability of several therapies and routes of administration, the selection and timing of appropriate treatments have become more complex. This article aims to review the therapeutic options for PAH and discuss optimum treatment strategies for their use.
Therapeutic Options for Pulmonary Arterial Hypertension
Calcium Channel Blockers
Calcium channel blockers (CCBs) are indicated in a small percentage of patients with idiopathic PAH who are acute responders to a vasodilators challenge during haemodynamic assessment. Inhaled nitric oxide (10–30 ppm) is the drug currently used in most centres, and a positive response has been defined as a decrease of mean PAP of at least 10 mmHg from baseline, with a drop of mean PAP below 40 mmHg and no decrease in cardiac output compared to baseline.7 Dosages of CCBs used in this setting are usually high: 120–240 mg for nifedipine, 10–20 mg for amlodipine and 240–720 mg for diltiazem.
Endothelin Receptor Antagonists
Endothelin-1 is overexpressed in several forms of pulmonary vascular disease and may play an important pathogenetic role in the development and progression of PAH.8 Two ERAs are approved for use in PAH: bosentan and ambrisentan. They are administered orally. Unlike bosentan, which is a sulphonamide class agent, ambrisentan is a propanoic acid class molecule. In vitro studies have shown that ambrisentan has a high binding affinity for the endothelin-A (ETA) receptor, with a selectivity for ETA versus endothelin-B (ETB),9 while bosentan exerts a dual receptor blockade. These drugs have demonstrated improvements in pulmonary haemodynamics, exercise capacity, functional status and clinical outcome in several randomised placebo-controlled trials.10–12 There is evidence to suggest that first-line bosentan therapy and the addition of another specific drug in case of clinical worsening improves survival in patients with advanced PAH.13 Similar data have obtained with ambrisentan.14 However, increases in hepatic enzymes to three-times the upper limit of normal have been observed in 11 % of patients treated with bosentan in clinical trials.15 Another ERA, sitaxentan, was developed and used in clinical practice but has been withdrawn.16
Phosphodiesterase-5 Inhibitors
The pulmonary vascular bed is targeted by numerous vasoactive factors including those that utilise cyclic guanosine monophosphate (cGMP) as an intracellular second messenger. These include nitric oxide and the natriuretic peptide family (atrial, brain and C-type natriuretic peptides). Phosphodiesterase-5 (PDE5) inhibitors inhibit cGMP metabolism and have been demonstrated to improve pulmonary haemodynamics and exercise capacity in patients with PAH.17 Two PDE5 inhibitors are approved for use in PAH – the oral therapies sildenafil and tadalafil. Data from RCTs suggest that these drugs have good short-term efficacy and are very well tolerated.18,19
Prostanoids
Prostanoids act by binding several specific receptors that activate an adenil ciclase, causing an intracellular increase in cyclic adenosine monophosphate. Published studies suggest that they exert their therapeutic action through vasodilatation, platelet inhibition and vascular remodelling.20 Epoprostenol (synthetic prostacyclin, epoprostenol [EPO]) was the first treatment introduced for the management of PAH. It was at first used as a bridge to lung transplantation and in the 1990s it was the first drug shown to improve haemodynamics, exercise capacity and survival in patients affected by PAH.21,22 It remains the only therapy to have demonstrated a survival benefit in an RCT.22
As a result of the short half-life of EPO in the circulation (<3 min), it must be administered by continuous intravenous (IV) infusion and requires referral to a specialist centre for initiation and management, since abrupt interruption of the infusion can cause rebound PAH with symptomatic deterioration and even death.23 Furthermore, owing to the chemical instability of one form of EPO (Flolan®; GlaxoSmithKline) at room temperature and neutral pH value, ice packs may be needed throughout the infusion period. A thermostable EPO formulation (Veletri®; Actelion Pharmaceuticals), which does not require cooling, has recently been approved for use by the US Food and Drug Administration (FDA).24
The practical difficulties of EPO have led to the development of prostacyclin analogues. Treprostinil may be administered at ambient temperature in a physiological solution, which enables it to be administered either subcutaneously or by IV infusion in patients for whom subcutaneous (SC) infusion is not tolerated. However, administration via the SC route is limited by infusion site pain, which can lead to discontinued therapy in some patients.25,26 Long-term studies suggest that IV EPO27,28 and SC treprostinil29 might improve survival in patients with idiopathic PAH. Iloprost may be administered by inhalation, although its short duration of action requires frequent administration (6–9 times daily),30–32 and also by IV infusion where its short-term efficacy is equivalent to that of EPO.33 Given the longer half-life of treprostinil and relative selectivity for the pulmonary circulation compared with iloprost, there was a strong rationale for developing an inhalable formulation.32 A recent Phase III trial demonstrated that among PAH patients who remain symptomatic on bosentan or sildenafil, inhaled treprostinil improves exercise capacity and quality of life and is safe and well tolerated.34
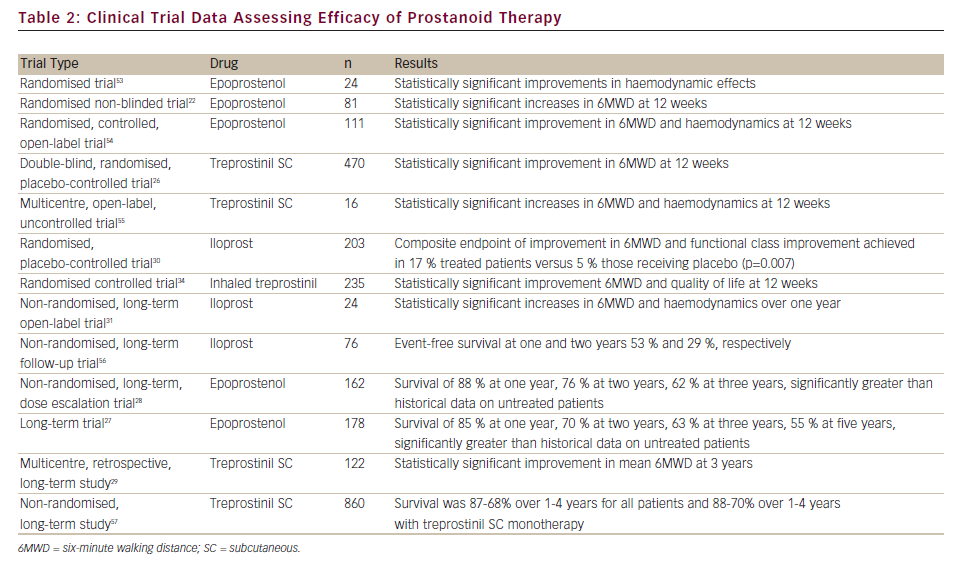
Furthermore, considerable research effort has been focussed on the development of an oral prostanoid for PAH therapy. Beraprost, an oral prostanoid,35 is approved in Japan and South Korea. Selexipag is an oral, long-acting prostacyclin receptor agonist pro-drug that was well tolerated and demonstrated efficacy in a recent Phase II clinical trial.36 Both are currently being evaluated in Phase III clinical trials. A summary of clinical trials and long-term studies demonstrating the safety and efficacy of prostanoids is given in Table 2.
Impact on Mortality
To date, all RCTs performed on PAH drugs have been designed to demonstrate the short-term clinical efficacy (six-minute walk test as the primary endpoint) of these drugs. Only in a few studies have there been a more robust endpoint of time to clinical worsening. Individual trials have been criticised for their endpoints of improvements observed on the exercise capacity, the short duration and small sample size. A meta-analysis of 16 RCTs in PAH concluded that the therapies produced limited benefits in clinical endpoints and failed to demonstrate a significant survival advantage.13 A more recent meta-analysis included 23 trials and found that although mortality remains high (3.8 % over the mean observation time of 14.3 weeks), there is a statistically significant reduction of 43 % in mortality after this time.37
Management of Treatment in Prostanoid Therapy
It is important to stress the need for adequate up-titration in order to have clinical efficacy in the use of prostanoids.26 Therapy is typically initiated at low dosage and increased to maximum tolerated dosage within weeks, as the drug escalation is limited by side effects such as flushing, nausea and hypotension. During long-term treatment, the patient may develop tolerance, identified by clinical deterioration and it becomes necessary to increase the dose over a period of weeks. It is also important to take into account the clinical state of the patient. Some centres (mainly in North America) adopt the strategy of increasing the dosage over time independently of the clinical status of the patients. In this setting, some patients develop a high cardiac output state that requires a reduction of the dose.38
Although the therapeutic range of prostanoids is high, in general, dosages fall within a range of 20–40 ng/kg/min with a trend towards higher dosage with treprostinil compared to EPO. There are isolated cases of patients on either drug being on very low doses or doses over 150 ng/kg/min. Regarding the dosage of treprostinil, in a recent retrospective review of 811 PAH patients it was found that a treprostinil dose of ≥40 ng/kg/min, and every 10 ng/kg/min dose increase, was associated with an improvement in long-term survival.39
The use of prostanoids is limited to expert centres because of the need for doctors and nurses experienced in the management of side effects, infection and parenteral administration. Administration by IV infusion carries a potential risk of infection, which can lead to life-threatening septicaemia.23 The short half-life of EPO leads to rapid deterioration or clinical pulmonary hypertension rebound, the patient running a higher risk of morbidity and potential mortality if drug delivery is interrupted owing to line or pump failure. Despite the need for an infusion system, the risk of infection and the adverse effects, the use of parenteral prostanoids should be encouraged, as these drugs are considered the most powerful ones in the treatment of PAH.40
Optimising Treatment Outcomes for Pulmonary Arterial Hypertension
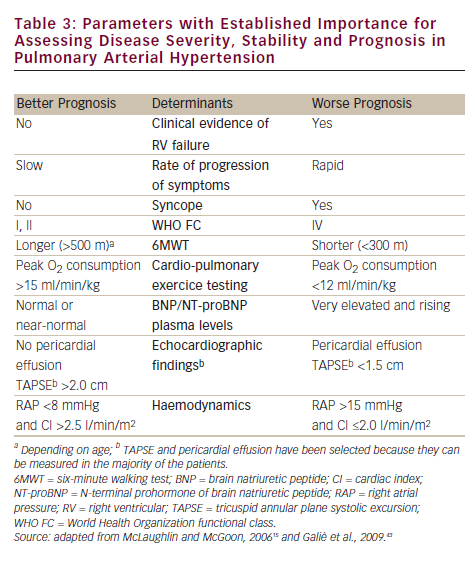
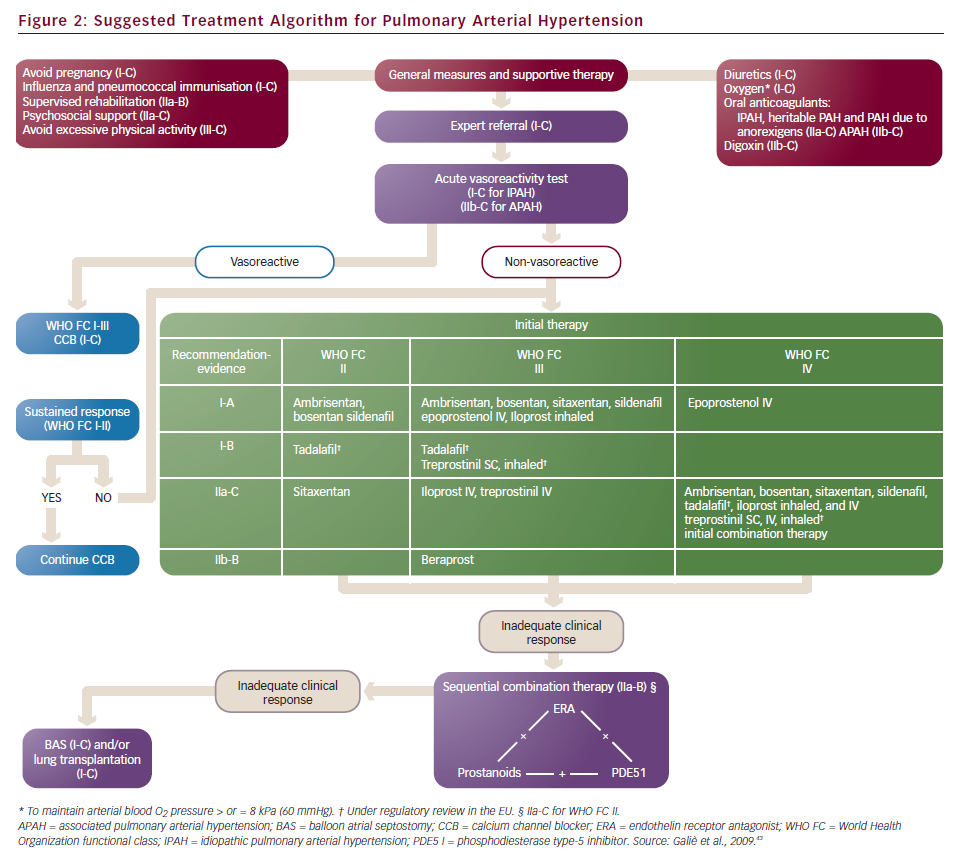
Until a few years ago, the treatment strategy in PAH was to start with monotherapy and add another drug in case of clinical worsening. Following some papers that suggested the poor efficacy of parenteral prostanoids when started in a very advanced stage (WHO FC IV),20,21,27,41 a more aggressive approach has been proposed: a goal-oriented strategy. Treatment goals in PAH therapy are those factors associated with a lower risk based on clinical evaluation (see Table 3), and treatment should be tailored to the individual. In agreement with a recent consensus document42 and guidelines,43 the current strategy is to start with one drug and re-evaluate the patients after 4–6 months, if the patient does not reach a predefined therapeutic goal another class of drug should be added to the first. A third class of drug may be added in case of inadequate clinical response. For patients initially in WHO FC II or III, this is a resulting clinical status defined as stable and not satisfactory or unstable and deteriorating. For patients who where initially in WHO FC IV, inadequate response is no rapid improvement to WHO FC III or better, or a resulting clinical status defines as stable and not satisfactory. Frequent follow-up is essential for patients commencing continuous infusion or inhaled therapy to ensure accuracy and compliance to therapy. A proposed treatment algorithm is given in Figure 2.43
Strict follow-up of PAH patients is a critical issue, as several studies proved that after an initial period of improvement, clinical worsening is a common experience among patients treated with oral drugs,13,44–46 leading to the need for oral combination therapy or parenteral prostanoid as an add-on therapy. The good survival rates reported in the long-term, open-label phases of the RCTs have created a misconception among non-expert centres that oral therapies represent a definite cure for PAH. A study of patients (n=821) taking bosentan as first-line therapy found that 90 % of PAH patients who died had not undergone any changes or additions to their therapeutic regimes. Several hypotheses have been proposed to explain this finding, including the possibility that a percentage of these patients had limited clinical documentation to support the addition or inclusion of prostacyclin therapy; that the prescribing physicians for this subset of patients were inadequately informed on the options for therapy escalation; and/or that the patients or physicians were aware of expert opinions but refused treatment changes due to perceptions regarding the complexity of parenteral prostanoid delivery.47
In a recent study it was demonstrated that patients on oral treatment have delayed referrals to expert centres for prostanoid therapy, and that this significantly impacts prognosis. Of the 57 patients who needed a parenteral prostanoid in an expert centre, non-survivors were more frequently referred from another hospital where they had started oral therapy (83 versus 36 %; p<0.01) and had a higher rate of urgent prostanoid treatment (69 versus 17 %; p<0.0001).48 Notably, patients who were referred from another hospital remained on oral therapy for a longer period than patients who had started oral therapy at the expert centre (850±600 versus 734±620 days), and the risk of death progressively increased in accordance with the modality of access to prostanoid therapy – lower in patients who had started oral therapy at the expert centre and higher in patients who had been referred from another hospital or needed first-line prostanoid therapy. These results emphasised the impact of late referral and the rapid progression of the disease on the fate of these patients.
This finding raises the question of whether the use of an oral therapy in a non-expert centre could delay the appropriate and timely use of parenteral prostanoids. In fact, most of the patients referred to the centre on oral therapy were in class IV and needed urgent prostanoid therapy. Supporting these conclusions is a recent paper that found that late initiation of IV iloprost in idiopathic IPAH patients who previously failed to respond to non-parenteral therapies was of limited efficacy in the majority of patients.49
As the guidelines take in the increasing number of results of RCTs, oral drugs are receiving a higher level of recommendation for patients in FC II and III, while parenteral prostanoids have been relegated to first-line use only in FC IV or for combination therapy. Considering the severity of the disease course, some expert centres are now suggesting the use of a more aggressive strategy. A recent registry study reported that first-line treatment of severe precapillary pulmonary hypertension with SC treprostinil is safe and efficacious over many years. If up-titration beyond six months is tolerated, effective doses are reached and outcomes are good.50
Findings from a recent open label study suggest that, in advanced cases, upfront combination tharapy (prostanoid and ERA) is associated with improvements in important outcomes such as functional class, exercise capacity and haemodynamics, and might favourably affect overall and transplant-free survival.51 The few clinical trials investigating the safety and efficacy of combined therapies have shown that they are generally well-tolerated – with mild but significant improvements in functional class, six-minute walk test and haemodynamics – but no long-term data exist and there remains the potential for additive effects and drug–drug interactions. A recent meta-analysis concluded that treatment of PAH with combination therapy improves multiple clinical and haemodynamic outcomes, but it does not reach the statistical significance for a 58 % reduction in mortality.52 Notably, the only RCT that has demonstrated a survival benefit of combination therapy was the Pulmonary arterial hypertension combination study of epoprostenol and sildenafil (PACES) study, which compared the combination of EPO plus sildenafil with EPO alone.53 Combination therapies are likely to be employed in patients displaying an inadequate response to single agents but it is not yet clear which regimens will be the most beneficial for which patients.
Conclusion
PAH is a serious and heterogeneous condition requiring careful monitoring and an individualised approach, and treatment should be managed by specialist PAH centres. Several therapeutic options are available, and advanced prostanoid therapy offers the unique ability to tailor the dose to an individual. This can present a challenge in terms of monitoring dosage, but the therapeutic range is wide and affords the flexibility to achieve a dose that can control symptoms and manage their side effects.
Despite considerable advances in treatment, the long-term survival of patients with PAH remains unsatisfactory. It is therefore necessary to optimise the usage of the existing therapies. The latter may be achieved by initiating prostanoid therapy at an earlier stage during treatment and ensuring adequate drug titration to maintain optimum therapeutic response. Future research should aim to better elucidate the pathogenesis of PAH, which remains incompletely understood, with the ultimate aim of developing curative therapies for this debilitating disease.