







Cardioprotection includes all methods and mechanisms that lead to the reduction in infarct size, and is thus involved in the evolution of post-ischaemic heart failure (HF). This is a growing research issue since ischaemic heart disease is the leading cause of morbidity and mortality worldwide. A key challenge for these studies is the unravelling of cardioprotection complexities such as the mechanisms leading to myocardial damage, the cascade of activated cellular signalling pathways, and the cellular and extracellular districts involved, which are strictly interconnected. This liaison between the heart and thyroid hormone (TH) adds complexity as it arises from the numerous TH effects on the heart, starting with cardiac differentiation in the transition from foetal to postnatal growth, during which THs induce transcriptional programming leading to the typical gene expression profile of the adult heart, as well as the maintenance of cardiovascular homeostasis directly through genomic and non-genomic actions and indirectly by TH regulating effects on other systemic pathways. Recent evidence highlights multiple actions of the TH system on the heart that can have a relevant role in cardioprotection, including the regulation of different intracellular pro-survival pathways, the preservation of mitochondrial function and morphology, the antifibrotic and proangiogenic effect, and also the potential induction of cell regeneration and growth. This review mainly focuses on these new actions of TH on the heart in relation to cardioprotection.
Definition of Cardioprotection
According to Heusch,1 cardioprotection is a highly concerted spatiotemporal programme in which different factors with different pathophysiological mechanisms are involved, at different times, in minimising irreversible ischaemic damage and favouring the functional recovery of the injured myocardium. In this context intracellular prosurvival pathways are activated in a complex cross-talking system. Taken as whole, cardioprotection can actually be defined as a complex dynamic network of cooperating units, which is characterised by global properties independent of the details of the units in the absence of cooperation.2 This suggests the need to take into account the intertwisted actions of the large number of components to understand the network of cardioprotection in order to give effectiveness to the therapeutic approaches. There are three main stages of myocardial damage:
- in the acute phase, coronary occlusion is the ’primum movens‘ of myocardial damage, causing ischaemic injury;
- this is followed by coronary revascularisation, i.e. percutaneous coronary angiography that causes reperfusion injury;
- in the chronic phase, when left ventricular dysfunction develops, the post-ischaemic remodelling process is another dynamic mechanism influencing myocyte function and survival.
Furthermore, the complexity of cardioprotection lies in the activation of the molecular mechanisms of cytoprotection, including activation of heat shock proteins (HSP), protein kinase C (PKC), extracellular signalregulated kinases (ERK), protein kinase B (AKT), p38 mitogen-activated protein kinase (p38MAPK), as well as the stimulation of cell growth, angiogenesis and metabolic adaptation. In addition, the maintenance of mitochondrial integrity is an emerging aspect of cardioprotection. Mitochondria participate in the regulation of myocardial calcium flux, myocyte cell death reactive oxygen species (ROS) generation and antioxidant response.3 The mechanisms of mitochondrial injury are different during ischaemic/reperfusion (I/R):
- The activation of the proapoptotic B-cell lymphoma 2 (BCL-2) proteins leads to mitochondrial outer membrane permeabilization, the release of cytochrome complex, caspase activation and apoptosis.4
- Oxidative stress can lead to a sudden increase in inner mitochondrial membrane permeability that is attributable to the opening of the so-called permeability transition pore (PTP), whose opening is accompanied by the release of ROS and calcium.5,6 This can culminate in the activation of calcium-dependent proteases (calpains) and lipases (cPLA2), inducing necrotic cell death.7,8
In the chronic phase, the activation of the neuroendocrine system, gathering renin-angiotensin-aldosterone and natriuretic peptides in the sympathetic autonomic nervous system, as well as prompting the inflammatory system, is initially protective and adaptive to haemodynamic changes induced by reduced cardiac output, conferring resistance to myocardial hypoxic injury.9 However, when these systems are continuously activated, their initial protective mechanisms become at first less effective, then maladaptive and dangerous for the entire body and heart, contributing to myocardial damage and progression of HF syndrome.10–12
Thyroid System in Patients with Acute Myocardial Infarction
In the clinical setting of acute myocardial infarction (AMI) the most frequent alteration of TH metabolism is low triiodothyronine (T3) syndrome.13 This occurs within 12 hours from the onset of symptoms, reaching the nadir at 72 hours. Low T3 syndrome is associated with a larger myocardial infarction (MI) and intense pro-inflammatory and stress response14,15 and, similarly to higher post-ischaemic levels of reverse T3, the TH inactive metabolite is considered an independent predictor of short-term and long-term mortality.16 Additionally, a large amount of clinical data support the clinical and prognostic role of altered thyroid metabolism in HF patients.17–22 T3 circulating levels were higher in patients with New York Heart Association (NYHA) class I and II with respect to patients in NYHA class III and IV with high brain natriuretic peptide (BNP) levels and lower left ventricular ejection fraction (LVEF).18,20,23
Thyroid System in Patients with Heart Failure
Low T3 syndrome and subclinical hypothyroidism have been associated with a worse prognosis in patients with HF. In particular, the prognostic power of low T3 syndrome was independent and additive with respect to conventional clinical and cardiac variables, such as LVEF. Furthermore, the negative prognostic power is enhanced in patients with higher BNP concentration both in acute decompensated and chronic compensated HF.23,24 Moreover, in patients with clinically stable HF, short-term synthetic T3 replacement therapy significantly improved neuroendocrine profiles and ventricular performance, characterised by an increase in stroke volume and reduction in the plasma circulating levels of noradrenaline, N-terminal pro-B-type natriuretic peptide (NT-proBNP) and aldosterone.25 According to the clinical data, experimental evidence shows that abnormal TH metabolic patterns, such as hypothyroidism and low T3 syndrome, can cause several histological, molecular and structural abnormalities within the myocardium that can be reversed after normalisation of the TH metabolic profile.26
Thyroid System and Cardioprotection
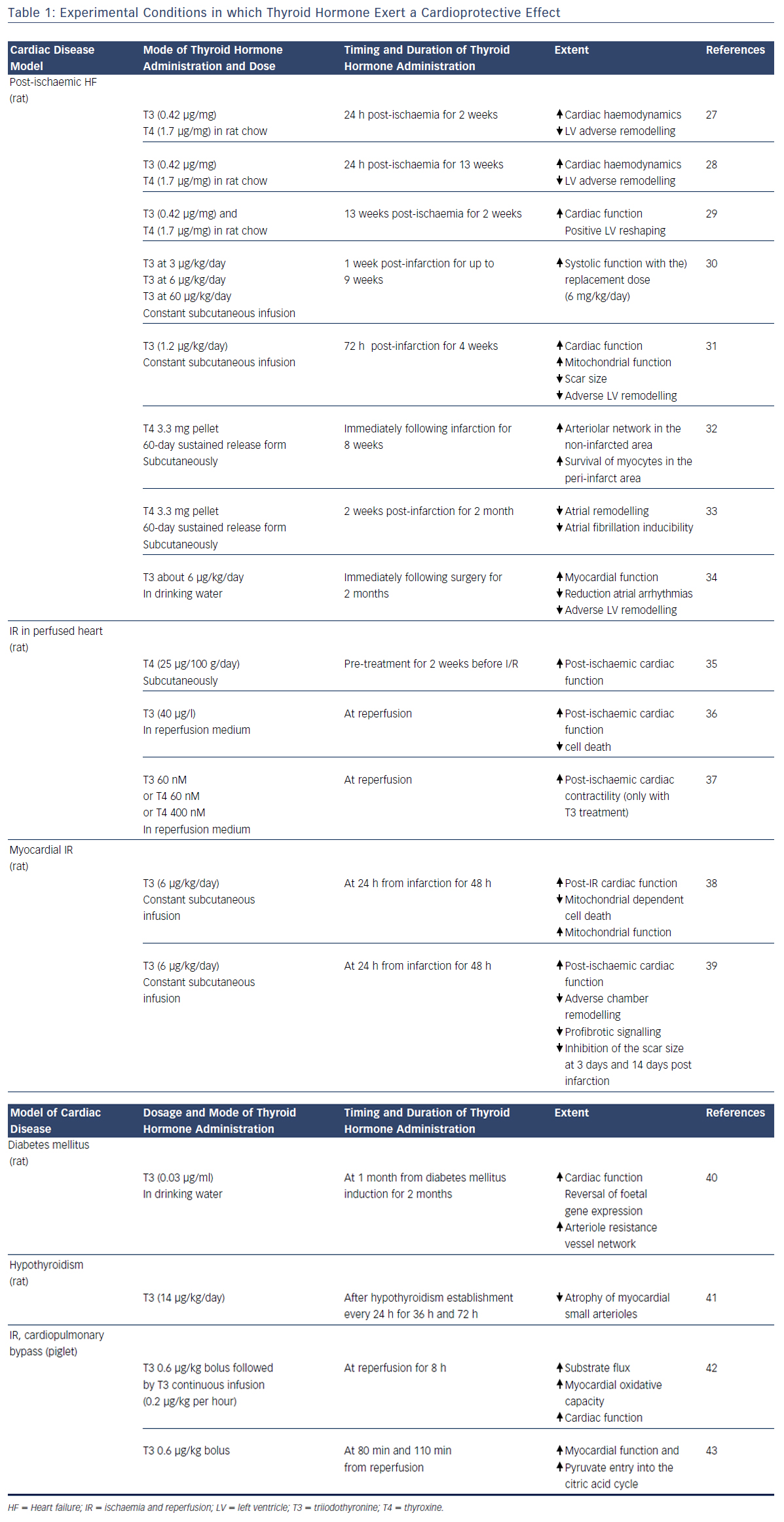
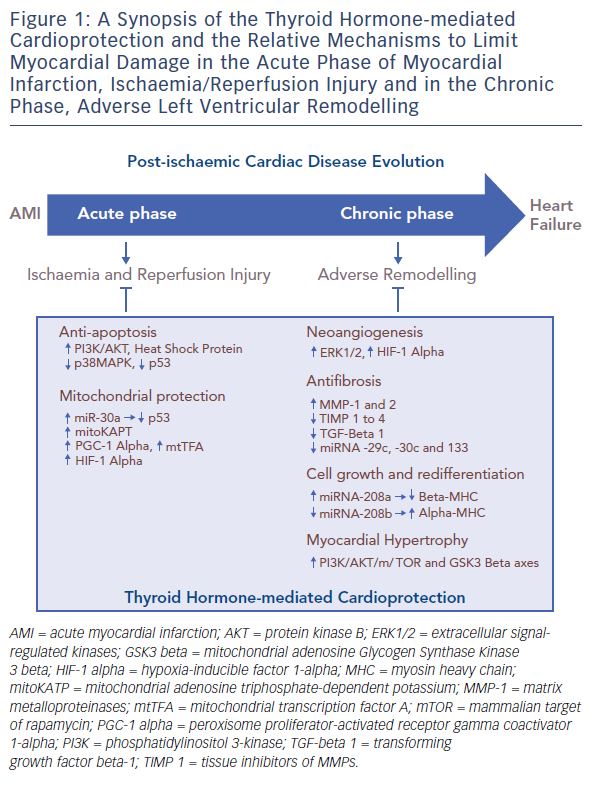
A large amount of experimental evidence highlights that TH can effectively play a role in the complex scenario of cardioprotection (see Table 1),27–43 and that this role is multifaceted due to the numerous actions and regulating mechanisms mediated directly or indirectly by the TH system (see Figure 1). In fact, TH-mediated actions are carried out at the level of the myocytes, the interstitium and the vasculature. TH also plays an important role in orchestrating the activation and function of different pro-survival intracellular pathways that are involved in cardioprotection. Furthermore, TH influences the neuroendocrine hormonal pathways activated in HF, as well as the inflammatory system.
Thyroid System and Pro-survival Intracellular Pathways
THs regulate the activation and function of the phosphatidylinositol 3-kinase (PI3K)/Akt and PKC signalling cascade, the expression, phosphorylation and translocation of HSP70 and HSP27;36,44 and the suppression of p38MAPK signalling.35 In particular, T3 treatment for 3 days after AMI reduced myocyte apoptosis in the border area of infarction via AKT signalling activation and also through decreased p38MAPK activation.32,36 Importantly, TH has a dose-dependent effect on AKT phosphorylation that can be mild45 and beneficial at low TH dose, while further induction of AKT signalling by higher doses of TH can be accompanied by increased mortality and activation of ERK, a kinase that has been associated with pathological remodelling.36 In addition, 2 weeks of thyroxine (T4) administration increased HSP70 expression and decreased p38MAPK activation in response to ischaemia, changes that closely resemble ischaemic preconditioning.36 The same treatment led to an increase in the basal expression and phosphorylation of HSP27, and earlier and sustained redistribution of HSP27 from the cytosolmembrane to the cytoskeleton-nucleus cellular fraction.35 Such changes might help to protect the myocardium against ischaemic insult, resulting in the improvement of post-ischaemic functional recovery.
Thyroid System and Mitochondria
The TH system has multiple actions to protect mitochondria. Among them, the TH is an important regulator of the tumour suppressor p53. This protein network is activated under stress conditions such as AMI and enhances the mitochondrial pathway of cell death.46 p53 expression is blunted by microRNA 30a (miR-30a) through direct targeting.47 In the post-ischaemic setting the miR-30a levels drop, which contributes to p53 accumulation, enhanced mitochondrial dysfunction and bcl-2-like protein 4 (BAX) activation, with the result of extended myocardial cell loss. In the post-ischaemic setting, T3 treatment counteracts the decrease in miR-30a levels, thus limiting the activation of p53 and the cascade leading to mitochondrial injury and cell death in the border zone of MI.38
Translationally, in patients with de novo post-ischaemic HF within 1 year of AMI, the levels of p53-responsive microRNAs (miR-192, miR-194 and miR-34a) were elevated in the early phase of AMI and were associated with increased left ventricular diastolic dimension.48 Moreover, T3 can protect mitochondrial integrity through a mitochondrial adenosine triphosphate-dependent potassium pathway, and by increasing the expression of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1 alpha) and the mitochondrial transcription factor A (mtTFA) in the border zone of infarction.31 These are key intracellular signals that control mitochondrial activity and biogenesis, and whose overexpression limits post-ischaemic left ventricular (LV) remodelling and preserves cardiac performance.49,50 Further T3 treatment preserves the expression of hypoxia-inducible factor 1-alpha (HIF-1 alpha), whose protective effect against reperfusion injury is mediated by inhibiting the mitochondrial opening of the PTP.7,8,51,52
Thyroid System and Foetal Recapitulation
During the evolution of HF, a foetal profile of gene expression is observed. This condition known as foetal recapitulation, is characterised by an isoform switch from the fast contracting alpha-myosin heavy chain to the slow beta-myosin heavy chain, by a decrease in sarco/ endoplasmic reticulum calcium ATPase (SERCA)/phospholamban (PLB) ratio, and also by the preference of glucose metabolism over fatty acids.51 At its onset in response to stress conditions, such as hypoxia or ischaemia and reperfusion injury, this mechanism may be beneficial to lower energy expenditure and oxygen consumption of the diseased myocardium. It is now understood that it may become maladaptive if continuously maintained.52,53 Altered expression of the TH receptor (TR) isoforms seems to contribute to the reactivation of the foetal transcriptional profile.54 Increased beta-myosin isoform expression, as well as impairment of calcium handling and cell contraction, were associated with the overexpression of unliganded TH receptor alpha 1 (TR alpha 1) in neonatal cardiomyocytes.55,56 Inhibition of T3 binding to TR alpha 1 prevented the differentiation of cardiac embryonic cells.57 Furthermore, phenylephrine administration in the absence of TH induced a switch of myosin isoform expression to a foetal pattern, which was associated to the redistribution of TR alpha 1 from the cytosolic to the nuclear compartment.57 The inhibition of the mammalian target of rapamycin (mTOR) signalling abolished this TR alpha 1 response and favoured cell atrophy.58 Recent evidence indicates that myosin isoform switching is under the control of a complex network of microRNA, including miR-208a and b and miRNA 499, that also presides over cardiac hypertrophy mechanisms.59 miR- 208 family b is a cardiac-specific miRNA enclosed within the myosin heavy chain (MHC) genes. Therefore, the expression of miR-208a and miR-208b is associated with the expression of the MHC alpha and MHC beta, respectively.59 T3 induces MHC alpha up-regulation and vice-versa downregulation of MHC beta.30 Deregulation of TH signalling in cardiac disease leads to inhibition of alpha-MHC and miRNA-208a expression, while in vitro treatment with THs significantly upregulates alpha-MHC and miRNA-208a and reduces beta-MHC and miRNA-208b expression, as well as miRNA-499.60 Based on this, physiological TH concentration seems necessary to guarantee adequate miRNA levels and to avoid foetal myosin isoform switching.
Thyroid System and Myocardial Interstitium
Besides cardiomyocytes, other myocardial cell types contribute to cardioprotection including fibroblast and endothelial cells that play a key role for the maintenance of cardiac architecture and function, and are involved in the pathophysiological evolution of HF. The function of these cells is critical for the synthesis and degradation of extracellular matrix components, as well as for the regulation of cell proliferation, migration, differentiation and apoptosis.61,62 In the post-ischaemic wound healing process, an interstitial remodelling occurs due to abnormal synthesis and deposition of collagen along with the dysregulation of matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitors of MMPs (TIMPs).63 It has been demonstrated that MMPs increase whereas TIMPs reduce their activity following MI. A temporal and spatial pattern of MMPs and TIMPs activation has been documented where MMP-1,2,3,7,9 activate early, and MMP-8,13 and 14 later following AMI.64 TIMPs 1–3 expression is also reduced in both the remote and border infarcted zone, whereas TIMP-4 is decreased only in the border infarcted zone.65 TH treatment with both T3 and T4 is paralleled to a reduction of the interstitial fibrosis in animal models of ischaemic and non-ischaemic HF, and this effect may be related, at least in part, to the effect of TH on MMP and TIMP activity.66,67 T3-dependent cardiac hypertrophy was not accompanied by interstitial fibrosis but an increase of MMP-2 and TIMP-2 expression was evidenced.67 Similarly, in rats, cardiac hypertrophy induced by T3 treatment has been associated with an increase of MMP-1 activity, a reduction of collagen I and III and a decrease in TIMP-l and 4 expression.68 More recently, in long-term T4 treated MI rats, a reduction of collagen deposition in the LV noninfarcted area has been reported along with a tendency towards increased MMP-2 and TIMPs-1–4 expression.32 The antifibrotic effect of T3 is further suggested by the evidence that early T3 replacement in a rat model of ischaemia/reperfusion reduced the scar size while improving long-term cardiac performance, and these effects were associated with the inhibition of the profibrotic transforming growth factor beta-1 (TGF-beta 1) signalling cascade and with the maintenance of the antifibrotic miRNA-29c, 30c and 133 expression.39
Thyroid System and Neo-angiogenesis
THs exert well-documented pro-angiogenic effects. Accordingly, chronic hypothyroidism is associated with rarefaction of small arterioles within the myocardium with consequent impaired coronary vasodilation. This alteration is reversed by T3 administration that prompts the proliferation of vascular smooth muscle cells, pericytes and endothelial cells.41,69 This pro-angiogenic action is mediated by several molecular mechanisms and starts as a non-genomic action at the plasma membrane of the endothelial cells through the interaction with the integrin alpha V beta 3.26 The transduction of the TH signal is driven by the mitogen-activated protein kinase ERK1/2 with the consequent transcription of pro-angiogenic genes, such as basic fibroblastic growth factor (bFGF) and vascular endothelial growth factor (VEGF).70 Another molecular cascade implicated in the T3 pro-angiogenic effect is triggered by the expression of HIF-1 alpha, which is induced by the activation of P13K signalling through the binding of TH with cytoplasmic TR beta.71,72 T3 induced angiogenesis has been documented in several experimental rat models of cardiovascular disease, including ischaemia, hypertension and diabetic cardiomyopathy.32,66,73 In a rat model of post-ischaemic HF, T3 supplementation to correct the low T3 state favoured a better retention of capillary density in the border zone in association with HIF-1 alpha stabilisation and TR alpha 1 upregulation.72
Thyroid System and Myocardial Hypertrophy
Post-ischaemic LV remodelling is the final result of molecular, subcellular, cellular and interstitial processes leading to changes in cardiomyocytes, extracellular matrix and vasculature within the peri-infarcted region (border zone) and remote region.74 Myocardial hypertrophy is one of the adaptive mechanisms involved in left ventricular remodelling and is influenced by TH dyshomeostasis. Hypothyroidism induces abnormal myocyte growth characterised by cell lengthening from the addition of sarcomeres in series, a change specific to dilated HF.75 In the study by Tang et al.,75 chronic hypothyroidism in rats, treated with propylthiouracil (PTU) for 1 year, caused progressive systolic dysfunction and heart dilatation associated with myocyte lengthening due to series sarcomere addition, which is a typical myocyte remodelling in HF76 Similarly, myocyte atrophy of myocytes characterised by an increase in length:width ratio occurred after 4 weeks treatment with PTU in rats. These changes were reversible 6 weeks after discontinuing PTU treatment.77
Conversely, TH treatment favours physiological hypertrophy through the activation of PI3K/AKT/mTOR and GSK3 beta axes, and through genomic regulation of specific target genes that encode both structural and functional proteins.78 As evidenced by a histological study, TH treatment of rats with hypertension and dilated HF, induced growth in myocyte transverse dimensions only, a change that reduced systolic wall stress.79 Furthermore, angiotensin type 1 receptor (AT1R) is a determinant of cardiac hypertrophy induced by hyperthyroidism, and also mediates TH induction of cardiac miR-208a and reduction of cardiac miR-208b levels in hyperthyroid rats. These data strongly suggest that AT1R might have an important regulatory role in cardiac muscle strength and contractility, influencing the efficiency of the cardiac function in hyperthyroidism.80
Thyroid System and Neuroendocrine Activation
In general, TH modulates the sympathetic and plasma reninangiotensin- aldosterone axis, and natriuretic peptide increasing their release and function.81 At the level of cardiac myocytes T3 promotes BNP gene transcription and regulates the cardiac betaadrenergic receptor-adenylate cyclase system by controlling the rate of transcription of the beta-1 adrenergic receptor gene. The net effect is the increase in BNP and catecholamines.82,83 In hyperthyroid rats the levels of norepinephrine in the cardiac muscle increased significantly, whereas it was undetectable in hypothyroid rats. Accordingly, increased and decreased NT-proBNP levels have been observed in patients with hyperthyroidism or hypothyroidism, respectively.84 Furthermore, in patients with HF and low T3 syndrome dobutamine infusion was associated with the TH metabolic normalisation and this, in turn, with short-term haemodynamic and neurohormonal improvement.85 Similarly, the same results have been obtained in the same patients through continuous L-T3 infusion for 3 days.25
Thyroid System and Inflammation
Plasma cytokines, in particular interleukin 6 (IL-6) and tumour necrosis alpha (TNF-alpha), are elevated in AMI and HF, and this is associated with the severity of the clinical status and a worse outcome.86,87 A cross-talk between TH and inflammation has been documented in experimental and human studies showing that administration of IL-6 in animals caused T3 decrease, due to the reduction in peripheral conversion of T4 into T3 following the inhibition of the 5’deiodinase activity.88–92
Conclusion
Cardioprotection can be considered an example of complexity applied to the human biological system, which is comparable to a non-linear, dynamic and intertwisted network in which small changes can result in important consequences and big changes may result in no or small consequences. In this complex dynamic network, the TH system may be a newly identified player orchestrating the different molecular, tissue and cellular elements involved both in the acute phase, when ischaemic/reperfusion injuries occur, and in the chronic phase, when the post-ischaemic remodelling process evolves. Notwithstanding the large amount of experimental data showing the potential effective role of TH on cardioprotection, there are still few clinical data available, in particular in the acute setting of AMI. It is important to underpin that the goal of TH treatment in patients with AMI and HF should be to restore and maintain euthyroidism in those patients with altered peripheral TH metabolism, and to avoid potentially dangerous pharmacological hyperthyroidism, as shown in two studies using TH replacement therapy in cardiac patients. The Coronary Drug Project (CDP), carried out in the early 1970s,93 demonstrated adverse outcomes, particularly with respect to the pro-arrhythmic effects of D-T4 (the inactive form of thyroxine). Patients were given 6 mg/ day D-T4, which is equivalent to 225 μg of L-T4, corresponding to more than double the endogenous production of T4, which is - 90–100 μg/day.94 It was later found that the D-T4 preparation used in the CDP was contaminated with a high level of active L-T4,95 which is the active form of thyroxine. Therefore, the cumulative dose of the D-T4 and L-T4 being administered was equivalent to several times the L-T4 dose that would be given to a patient to correct overt hypothyroidism. Furthermore in the study by Goldman et al., the TH analogue 3,5 diiodothyropropionic acid (DITPA) was administered to patients with HF.96 No improvement on outcome was observed, but rather fatigue was more frequent in the DITPA group, in association with weight loss, reduction in serum cholesterol and increase in heart rate, all signs and symptoms suggesting thyrotoxicosis. Therefore, several questions need to be answered, including: the type of hormone to administer, T3 or T4 or both; the dosage and duration of the treatment to reach the goal that is the restoration of euthyroidism; the starting point of the treatment, for example before, during or late after the revascularisation procedure; the patient selection, i.e. patients with altered TH metabolism; and finally the clinical, functional and prognostic targets to assess the effective benefit of TH treatment.