










Over the past decade, triglyceride-rich lipoproteins (TRLs) and their remnants have emerged as novel potential targets for managing dyslipidaemia and atherosclerotic cardiovascular disease (ASCVD).1 Conclusive clinical evidence has linked LDL cholesterol (LDL-C) with the risk for ASCVD. Numerous studies in a variety of patient populations have established that statin therapy promotes robust reductions in LDL cholesterol (LDL-C) and significantly reduces the risk of ASCVD.2 Guidelines throughout the world recommend statins for primary and secondary ASCVD prevention.3 However, despite statin intervention, approximately 60–70% of events in clinical trials are not prevented, partly due to suboptimal LDL-C reduction and elevated triglyceride (TG) levels.4 Clearly, residual risk remains unacceptably high. In addition, growing evidence supports the conclusion that non-HDL cholesterol (non-HDL-C), which includes TRLs and all apolipoprotein B (ApoB) lipoproteins, is significantly associated with ASCVD independent of LDL-C, which may explain a portion of ASCVD residual risk.5
Hypertriglyceridaemia, defined as a TG level of ≥150 mg/dl (≥1.7 mmol/l), is an exponentially growing cardiovascular (CV) risk factor.6 Moderate hypertriglyceridaemia is independently associated with ASCVD. It is linked with multiple CV risk factors, including obesity, metabolic syndrome, type 2 diabetes, fatty liver disease and epicardial steatosis, a variety of genetic polymorphisms, and chronic kidney disease.6 Severe hypertriglyceridaemia, defined as TG levels ≥500 mg/dl (≥5.6 mmol/l), is a known risk factor for acute pancreatitis and should prompt therapeutic intervention with TG-lowering medication.7
Plasma TG levels have been found to correlate closely with those of TRLs and other lipoprotein derangements, such as elevations in very LDL (VLDL) particles, intermediate-density lipoprotein (IDL), apolipoprotein C-III (ApoC-III), small dense LDL particles and low HDL-C, all of which are associated with an increased risk of CV disease (CVD).8 However, CV studies on TGs and, consequently, TRLs have yielded conflicting results, implying some uncertainty about the direct residual causal role of TRLs in the progression of ASCVD.6 Here, we provide an overview of the available epidemiological, genetic, and clinical evidence regarding the atherogenicity of TRLs and their role in CVD progression. We also review ongoing work with established and emerging therapies in development that target TGs and TRLs, and how they impact risk for ASCVD events.
TG-rich Lipoprotein Metabolism
TG and cholesterol esters are essential circulating lipids.9 Given their hydrophobic nature, they require a lipoprotein carrier to be transported in the circulation and assist with their metabolism. Within hepatocytes, TG and cholesterol esters are combined with phospholipids, ApoB and free cholesterol to form VLDL.10 The TG and esterified cholesterol are carried within the hydrophobic core of the lipoprotein particles, sequestered from the hydrophilic milieu of blood. Multiple lipoproteins transport TG in the circulation, especially chylomicrons, VLDLs and their remnants.11 Remnant lipoproteins are lipoproteins, such as small VLDL and intermediate-density lipoprotein, whose triglyceride cargo are incompletely hydrolysed. Lipoprotein metabolism requires the participation of many apolipoproteins (Apo), which either allow lipoproteins to bind to cell surface receptors (ApoB, ApoE), align them sterically to interact appropriately with enzymes (ApoC-II) or inhibit enzymatic lipolysis (ApoC-III).12 TRL metabolism requires multiple steps and can occur via the exogenous and endogenous metabolism pathways. These pathways are summarised schematically in Figures 1 and 2.
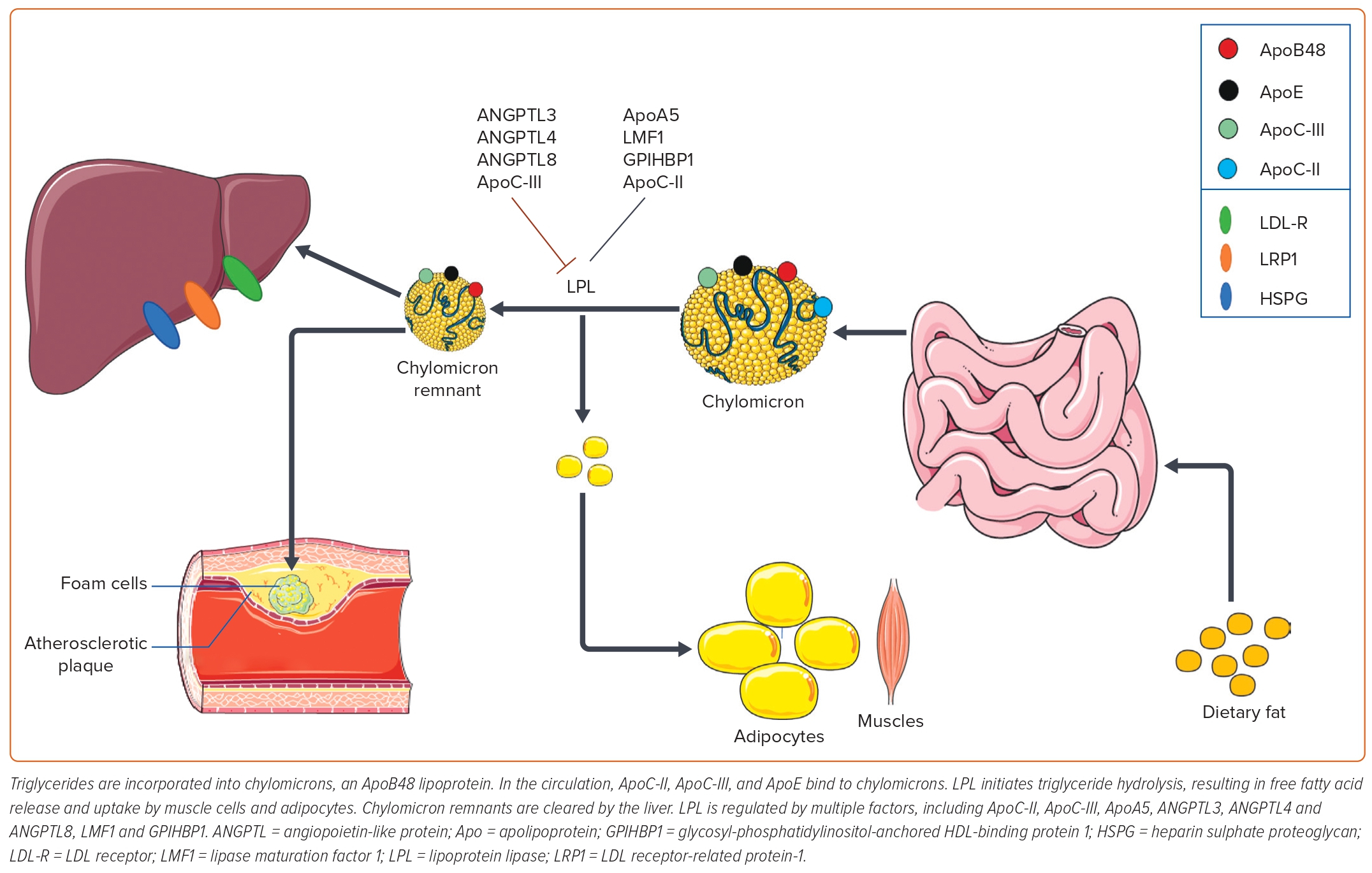
Exogenous Pathway
The formation of TRLs begins as a response to dietary fat absorbed by jejunal enterocytes. TGs are incorporated into chylomicrons within the enterocytes, which are large ApoB-48 (a truncated form of ApoB100)-bound TG-predominant lipoproteins.13 Chylomicrons enter the circulation via perimesenteric lymphatics and the thoracic duct. HDL is a reservoir of apoproteins and serves as an apoprotein donor to other lipoproteins to regulate or alter their functionality. While in circulation, HDL donates ApoAI, ApoC-II, ApoC-III and ApoE to chylomicrons. ApoC-II facilitates chylomicron binding to lipoprotein lipase (LPL), which then initiates triglyceride hydrolysis at the luminal surface of capillaries resulting in free fatty acid (FFA) release and systemic tissue uptake. The FFA can undergo oxidation via β-oxidation, be converted to glucose via phosphoenolpyruvate carboxykinase, reassembled into triglycerides and stored in adipose tissue, or loaded into VLDL and secreted into the circulation. LPL is regulated by multiple factors, including ApoC-III, ApoA5, angiopoietin-like proteins 3, 4 and 8, and other factors discussed in the genetics section of this review.14 As the chylomicron is more completely lipolyzed, it forms chylomicron remnant particles of various sizes. Chylomicron remnants are cleared by the liver via the LDL-R and the LDL receptor-related protein-1 (LRP-1), which is an ApoE-binding receptor found within hepatic sinusoids.15 LRP-1 is also the principal receptor responsible for clearing VLDL remnants and IDLs from circulation.
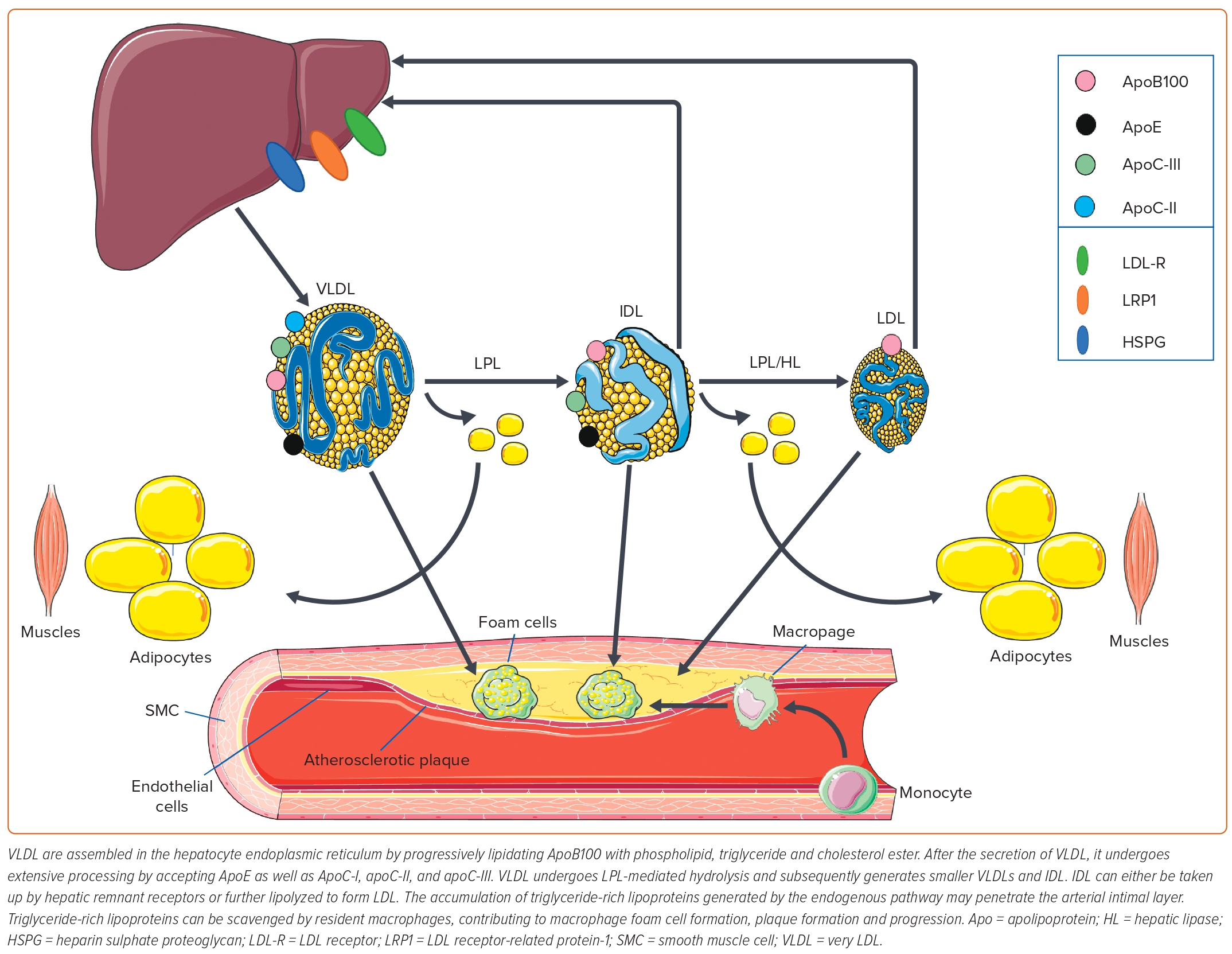
Endogenous Pathway
The endogenous pathway occurs within hepatocytes and is defined by VLDL biosynthesis. VLDL is assembled in the endoplasmic reticulum by progressively lipidating an ApoB100 scaffold with phospholipid, triglyceride and cholesterol ester. VLDL TG is derived from recycled fatty acids taken up from plasma by remnant-like particles (RLPs) as they return to the liver or fatty acids synthesised de novo in hepatocytes. After the secretion of VLDL, it undergoes extensive processing by accepting ApoE, as well as ApoC-I, ApoC-II and ApoC-III donated by HDL. Subsequently, VLDL undergoes LPL-mediated hydrolysis, generating smaller and denser VLDL particles. Small VLDLs can be further lipolysed into IDL. IDL can either be taken up by hepatic remnant receptors or can be further lipolysed to form LDL particles, the end-product of ApoB100-containing lipoprotein metabolism.15 Importantly, the accumulation of remnant lipoproteins generated by either pathway is secondary to increased TRL production, impaired lipolysis in plasma, decreased clearance caused by multiple genetic variants affecting TRL metabolism and hepatic uptake or all of these mechanisms depending on the metabolic milieu. Each of these mechanisms for generating altered TRL metabolism are strongly associated with increased ASCVD risk.
Proposed Pathophysiology of TG-rich Lipoproteins in the Progression of Atherosclerosis
It has been proposed that the cholesterol content of the TRL remnants primarily contributes to the progression of atherosclerosis rather than the TG.9 Similar to LDL particles, cholesterol-rich and TG-depleted TRL remnants can penetrate the arterial intimal layer, where they selectively bind to the connective tissue matrix (collagen, elastin, laminin, fibronectin etc.). Once embedded in the subendothelial space, TRLs can be scavenged by resident macrophages, contributing to macrophage foam cell formation, plaque formation and atherosclerosis disease progression.16 TRL are thought to exert as much or more proatherogenic influence as LDL particles. Unlike LDL, the remnants of TRL can be scavenged directly by arterial macrophages without oxidative modification by such enzymes as myeloperoxidase or 15-lipoxygenase.17 Because of their larger size, they can carry more cholesterol per particle than LDL.16 Furthermore, studies have also demonstrated that TRL remnants induce endothelial dysfunction, stimulate inflammation and promote atherogenesis.18
LPL-mediated TRL hydrolysis forms and releases a variety of bioactive lipid products, such as oxidised FFA, along the luminal surface of capillaries. These lipolytic products and TRLs are also known to activate multiple inflammatory and pro-oxidative signalling pathways, coagulation, and apoptosis, which play fundamental roles in atherogenesis.16 It has been shown that oxidised FFAs increase the expression of inflammatory interleukins and cytokines by infiltrating white blood cells, such as neutrophils, T cells and monocytes, promoting arterial wall inflammation.19,20 Simultaneously, TRL remnants upregulate the expression of intercellular adhesion molecule-1, vascular cell adhesion molecule-1 and a variety of selectins on the endothelium.21,22 These adhesion molecules possess proatherogenic and pro-inflammatory properties that enhance permeability and promote transendothelial migration of leukocytes (monocytes that subsequently convert to resident macrophages, T helper cells and mast cells) into the subendothelial space where they create an inflammatory nidus.23 Endothelial activation mediated by TRL remnants leads to increased white cell adhesion and inflammatory responses.21,22 TRL remnants have also been reported to induce early activation of neutrophils.24
TRL remnants increase the production of reactive oxygen species (these include superoxide anion, hydroxyl radical and peroxynitrite), which may increase endothelial permeability and toxicity, which promotes leukocyte adhesion, cell injury and death at high concentrations.20 TRL remnants are also known to induce apoptosis of endothelial cells through increased secretion of pro-apoptotic cytokines, tumour necrosis factor-α and interleukin-1β. This process contributes to progressive vascular injury and atherosclerosis.25 TRLs and their remnants promote platelet aggregation and coagulation by facilitating the assembly of the prothrombinase complex and upregulating the expression of plasminogen activator inhibitor-1 antigen.26 TRLs and their remnants have also been shown to upregulate endothelial expression of tissue factor, an essential initiator of the coagulation cascade.21,22 Finally, TRL remnants reduce HDL’s antiatherogenic and anti-inflammatory effects, and are significantly correlated with impaired coronary vasoreactivity secondary to reduced availability of nitric oxide.27
Factors required for TRL metabolism have also been reported to be associated with ASCVD, particularly ApoC-III, an inhibitor of LPL.12 ApoC-III promotes the progression of atherosclerosis through multiple mechanisms: it interferes with VLDL binding to hepatocyte ApoE receptors, promotes the formation of small and dense LDL particles, induces expression of pro-inflammatory mediators, and stimulates monocyte activation and adhesion of monocytes to endothelial cells.16,28 It has also been reported that ApoC-III induces apoptosis and impairs HDL function and its atheroprotective effects.29,30
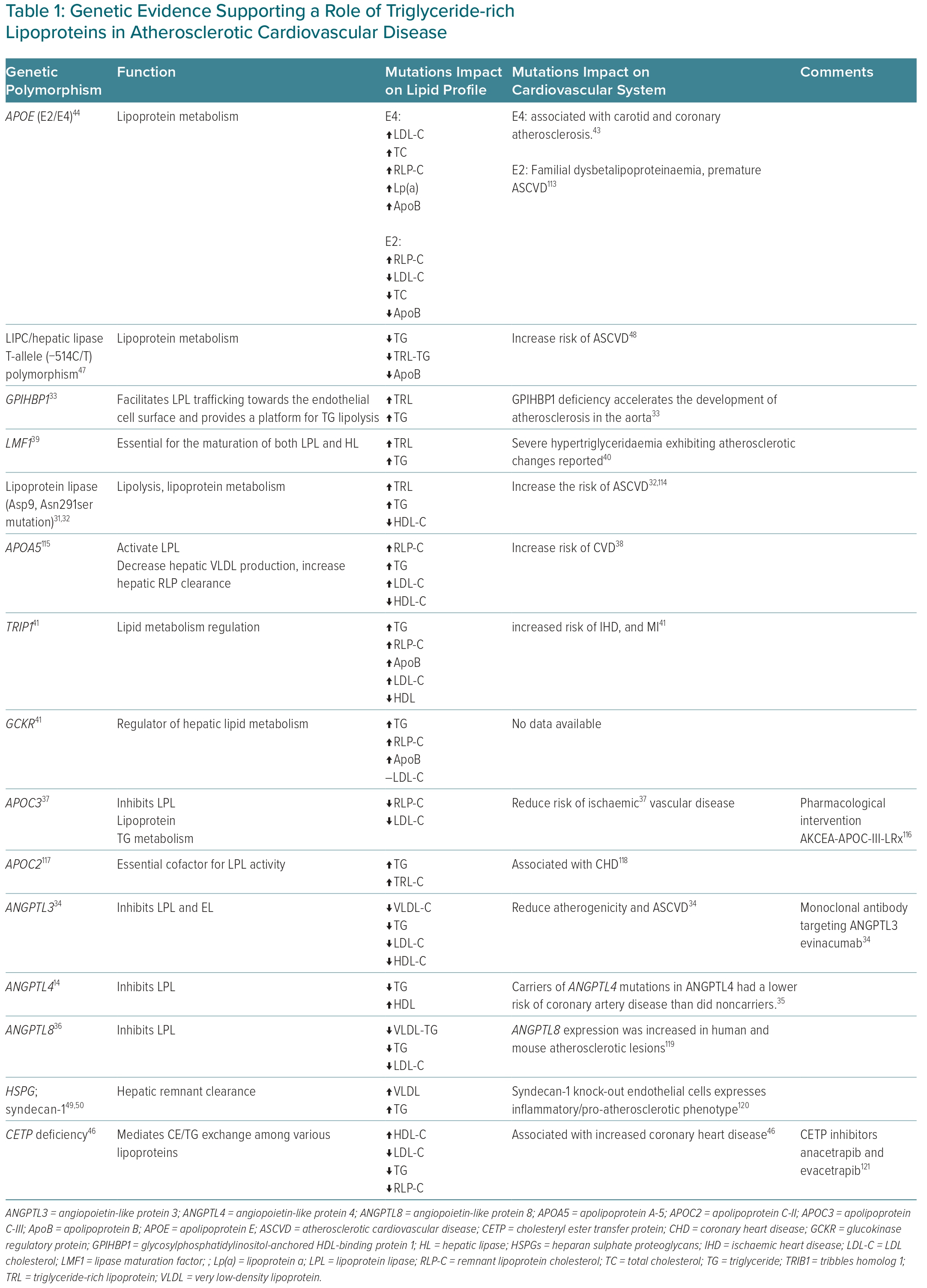
Genetic Evidence
Recently published data showed that multiple molecular pathways target RLP cholesterol (RLP-C) metabolism, the majority of which involve LPL function (Table 1). LPL, an enzyme that regulates TG hydrolysis in plasma, has multiple regulators impacting its activity.13 Genetic variations in LPL, such as the loss of function Asp9Asn and Asn291ser gene variants, are associated with elevations in TG and TRL-C, as well as increased ASCVD risk.31,32 Apart from direct mutations in the LPL gene, multiple genes have been shown to affect LPL indirectly. Glycosylphosphatidylinositol-anchored HDL-binding protein 1 is a protein that facilitates LPL trafficking towards the endothelial cell surface and provides a platform for TG lipolysis. Glycosylphosphatidylinositol-anchored HDL-binding protein 1 deficiency has been reported to affect TRL metabolism by increasing its levels and accelerating atherosclerosis and plaque instability in diabetic LDL receptor knockout mice.33
Angiopoietin-like protein 3 (ANGPTL3) is a member of the angiopoietin family and is associated with ASCVD.34 It has been studied as a potential target for managing TRL abnormalities. Although ANGPTL3 has been found to bind strongly to LDL receptors, the role of this interaction in atherosclerosis remains poorly understood. ANGPTL3 inhibits LPL and endothelial lipase, and its deficiency is associated with reduced VLDL, LDL-C and HDL-C levels. Moreover, other members of the angiopoietin family, including angiopoietin-like protein 4 and angiopoietin-like protein 8, are found to be physiological inhibitors of LPL and promote TRL elevation.35,36
An important factor that regulates TRL metabolism by inhibiting LPL is ApoC-III. Heterozygosity for ApoC-III loss of function mutations decreases the levels of RLP-C by 43% and decreases the risk of ischaemic vascular disease by up to 41%.37 Another factor that regulates LPL is ApoA-V, an activator of LPL, which increases TG lipolysis and RLP-C metabolism. Studies show that deficiency of ApoA-V secondary to loss of function mutations is associated with sex-specific changes: 1131C female allele carriers have significant associations with TRL and ASCVD, with a hazard ratio of 1.85 (95% CI [1.03–3.34]; p=0.04); in contrast, male 56G carriers show significant associations between ASCVD and elevated RLP.38
Lipase maturation factor 1 has revised our current understanding of LPL maturation. The nonsense mutation of (Y439X) in lipase maturation factor 1 is associated with severe hypertriglyceridaemia and lipase deficiency, indicated by the accumulation of TRL in a mouse model.39,40 Another factor associated with TRL is Tribbles homolog 1. Tribbles homolog 1 has a regulatory role in mitogen-activated protein kinase. The Tribbles homolog 1 rs2954029 polymorphism (chromosome 8, position 126,560,154 on the forward strand) is associated with higher levels of RLP-C and TG, and increased risk of ischaemic heart disease (IHD) and MI.41 Glucokinase regulatory protein is a protein that regulates glucose metabolism. The glucokinase regulatory protein rs1260326 and glucokinase regulatory protein rs780094 (chromosome 2, position 27,594,741 on the forward strand) polymorphism is associated with increased RLP-C and TG, without a significant association with ASCVD.41
One of the important key regulators of lipoprotein metabolism is ApoE.42 ApoE polymorphisms are associated with elevated TRLs, especially isoforms E2 and E4, and with premature ASCVD.43 Additionally, familial dysbetalipoproteinaemia (remnant removal disease) is a genetic disorder linked to the E2/E2 genotype.44 It is characterised by impaired clearance of remnants, which can lead to formation of atherosclerosis and increase risk of CVD. The Framingham Heart study found that individuals with the E2/E2 genotype had a significantly higher risk of developing ASCVD.45
Plasma cholesteryl ester transfer protein promotes the exchange of cholesteryl esters from HDLs to ApoB-containing lipoproteins. Cholesteryl ester transfer protein deficiency has been reported to increase HDL and significantly reduce RLP-C in heterozygous participants.46 Hepatic lipase (HL) is important in the metabolism and clearance of RLP. A hepatic lipase gene (LIPC) mutation in the promotor region (−514 C to T) has been reported to decrease HL and, subsequently, the catabolism of RLP-C.47 HL polymorphism has also been found to be associated with increased ASCVD risk.48 The clearance of RLP is orchestrated by multiple key regulators, including ApoE, HL, LPL and receptors expressed on the hepatocyte surface, such as the LDL receptor, LRP1 and heparan sulphate proteoglycan receptors found in the space of Disse.
Heparan sulphate proteoglycan receptors are attached to cell membranes and are composed mainly of syndecan 1 as a core protein. In vivo studies using heparinase to degrade heparan sulphate decreased the clearance of VLDL remnants. Moreover, introducing a mouse-specific knockout model (Ndst1−/−) to decrease the sulphation of HPSG increased the levels of TRL. A syndecan 1 knockout mouse model (Sdc1−/−) showed increased TG, as expected, due to the accumulation of TRL.49 Finally, heparan sulphate 2-O-sulphotransferase 1, an enzyme responsible for generating sulphated forms of proteoglycans, and its deficiency are associated with increased TG due to remnant lipoprotein accumulation.50 In conclusion, RLP metabolism and clearance are regulated by multiple complex molecular pathways that are as yet incompletely understood. The signalling mechanisms responsible for RLP metabolism will likely yield novel pharmacological interventions to potentiate RLP metabolism and reduce CV risk.
Epidemiological Evidence
The association between remnant lipoproteins and CV risk has been evaluated in numerous studies performed around the world (Supplementary Table 1). A landmark study from the Montreal Heart Institute of patients with pre-existing ASCVD showed that TRL remnants are associated with progression of coronary artery disease (CAD) independent of LDL-C.51 Multiple studies confirmed the association between TRL remnants and CV risk. The Framingham Heart Study reported that RLP is an independent CV risk factor in women.52 A logistic regression analysis showed that RLP-C was significantly associated with ASCVD, with a reported HR of 2.27 (95% CI [1.37–3.77]). In addition, the Honolulu Heart Study showed that RLP independently predicts the incidence of ASCVD, but only if it is associated with elevated TG levels in Japanese-American men.53 The study found a significant association between RLP-C and RLP-TG levels, and the relative risk of developing CHD, with corresponding p-values of 0.0022 and 0.0045, respectively.
The association between RLP and ASCVD was extensively studied and confirmed in the Danish population using data from the Copenhagen City Heart Study, Copenhagen General Population Study and Copenhagen Ischemic Heart Disease Study. This analysis showed a causal association between RLP and increased risk of MI, with an odds ratio of 1.87 (95% CI [1.25–2.81]) in patients with higher levels of RLP.54 This study was followed by a series of studies by Varbo et al. to expand the evaluation of the impact of RLP on ASCVD in the Danish population. The first study showed that elevated postprandial RLP-C was associated with a 2.8-fold causal risk increase of IHD independent of reduced HDL-C.55 The second study confirmed the first study’s findings and showed that elevated postprandial RLP-C is causally associated with IHD with a causal risk ratio of 3.3 (95% CI [2.1–5.2]).56 This study also revealed the dual atherogenic and inflammatory properties of RLP-C compared with the atherogenic effects of LDL-C (RLP-C was approximately 10-fold more pro-inflammatory than LDL-C). The third study further confirmed the impact of RLP on ASCVD and its association with other CVD risk factors, such as hypertension, obesity and dyslipidaemia.57 This study reported that RLP-C, in combination with LDL-C and elevated blood pressure, partly mediated IHD in obese participants. Moreover, in another study of the Danish population, elevated RLP-C was associated with IHD, MI and increased all-cause mortality risk.58
Combined data from the Framingham Offspring Cohort Study and the Jackson Heart Study examining a total of 4,932 participants showed that RLP-C levels predict the incidence of CAD, with a hazard ratio of 1.23 per 1-SD increase (95% CI [1.06–1.42]).59 This association was not as strong as reported in the Danish population, which may reflect population-dependent differences. Data from the Atherosclerosis Risk In Communities study showed that RLP-C was weakly associated with CAD and ischaemic stroke risk only in minimally adjusted analyses.60 A recent study using data from the Copenhagen General Population Study and Copenhagen City Heart Study studies reported a robust, consistent association between RLP-C and peripheral artery disease (PAD), MI and ischaemic stroke, with a HR as high as 4.9 for peripheral artery disease.61
The Multi-ethnic Study of Atherosclerosis found that nuclear magnetic resonance spectroscopy-measured RLP-C was independently associated with ASCVD events, with a HR of 1.20 (95% CI [1.04–1.39]).62 Finally, data from different populations and continents, including data from Japan, South Korea and China, have reported an association between RLP-C and ASCVD.63–65 In conclusion, these studies suggest that TRLs and their remnants are an integral risk factor for ASCVD; the differences in hazard ratios are likely related to differences in populations studied and use of different separation methodologies for measuring various remnant lipoproteins and their subclasses.
Controversial Aspects of TG-rich Lipoproteins and CVD
There is an ongoing controversy regarding the differences between TRLs types and subclasses, and their impact on CVD. One of the controversial aspects is questioning whether TRLs are causally associated with the development of ASCVD or if they are just markers of other underlying risk factors for ASCVD. For instance, several studies reported that TRLs are causally associated with an increased risk of ASCVD.54,55 In contrast, other studies showed an attenuated association of RLP-C and ASCVD after adjusting for multiple confounders.60 Moreover, controversy has arisen over the identification of specific TRL subclasses, their measurement and their impact on ASCVD. Several studies pointed out that the size and density of TRLs affect ASCVD risk differently. For example, a recent meta-analysis showed that smaller-density TRLs have a more significant association with ASCVD than larger TRLs.66
Measuring TRLs using different techniques raises controversies over the association of TRLs with ASCVD. A study conducted among participants of the Copenhagen General Population Study found that both directly measured and calculated RLP-C were associated with an increased risk of MI. However, the association was found to be stronger for directly measured RLP-C compared with calculated RLP-C.67 This study also reported that a significant number of individuals with high levels of directly measured RLP-C were not identified as high-risk by the calculated method, suggesting that they would have been overlooked as being at increased risk for MI.
Differences in measurement and the lack of standardisation for measuring RLP have affected the comparability across the RLP and ASCVD studies. Several studies used calculated measurements, whereas other analyses used gel electrophoresis, nuclear magnetic resonance spectroscopy, ultracentrifugation, and other techniques of separating and measuring TRLs. Finally, to minimise inconsistent findings and controversy over the role and mechanism of TRLs in ASCVD, further effective standardised strategies are needed to better understand the nature of the relationship between ASCVD risk and TRLs.
Available Treatment Options to Reduce TG and TG-rich Lipoprotein Levels
Guidelines define the severity of hypertriglyceridaemia differently.8,68 However, the guidelines define severe hypertriglyceridaemia as a TG level of ≥500 mg/dl (≥5.6 mmol/l), and they recommended initiating TG-lowering therapy to decrease the risk of pancreatitis.69 Although the guidelines do not specify a target TG level to achieve, they emphasise the importance of lifestyle modifications as the first line of therapy to reduce CV risk. If TG and TRL remain elevated despite lifestyle intervention and statin therapy, guidelines recommend using TG-lowering therapeutics, primarily fibrates or Ω-3 fatty acids.6,8,68,69 Patients with hypertriglyceridaemia have shown a significant decrease in CVD risk when treated with eicosapentaenoic acid (EPA) in addition to statin therapy. The REDUCE-IT trial provided the first evidence of a reduction in CV mortality when EPA was added to background statin therapy.70 Although fenofibrate will help to reduce TG further, there is no evidence, however, that the addition of fenofibrate in this scenario provides incremental ASCVD risk reduction.
Fibrates
Fibrates reduce the levels of TG by approximately 36%, non-HDL-C by 6–16% and LDL-C by 8%, and increase HDL-C by approximately 10%.71,72 Several clinical trials and meta-analyses have examined the impact of fibrates on CV outcomes. The Helsinki Heart Study included 4,081 asymptomatic middle-aged men with a documented history of primary dyslipidaemia and showed that gemfibrozil reduced CV events by 34% (95% CI [8.2–52.6]; p<0.02).73 In the VA-HIT trial, 2,531 men with established ASCVD were randomised to receive either gemfibrozil 1,200 mg/day or placebo. Gemfibrozil therapy reduced the risk for the primary composite endpoint by 22% (95 CI% [7–35]; p=0.006) relative to placebo.74 Gemfibrozil is not widely used because the two aforementioned trials were not performed against a statin background, and gemfibrozil is not safe to use in combination with a statin. Gemfibrozil inhibits the glucuronidation of the statins and can increase risk for myopathy and rhabdomyolysis.75
Type III hyperlipidaemia is characterised by elevated levels of VLDL remnant lipoproteins and is associated with a high risk for ASCVD. In a group of 12 patients with type III hyperlipidaemia, gemfibrozil therapy reduced VLDL cholesterol and TG by 72% and 68%, respectively, when dosed at 600 mg per oral twice daily.76 Using an immunoaffinity separation technique, Karpe et al. showed that gemfibrozil therapy reduced RLP-C by a median of 34% and that elevations in RLP-C were associated with both coronary and saphenous vein graft atherosclerotic disease progression.77 In a study using density gradient ultracentrifugation, among type 2 diabetes patients given a fat load, gemfibrozil reduced VLDL1, VLDL2 and IDL by 38, 22 and 5%, respectively.78
In the Bezafibrate Intervention Project, 3,090 participants with a previous history of MI or stable angina with dyslipidaemia were randomised to receive either 400 mg of bezafibrate per day or a placebo.79 In a post hoc analysis in the subgroup of patients with high TG (>200 mg/dl), bezafibrate was significantly associated with a 39.5% (p=0.02) reduction in the cumulative probability of fatal or nonfatal MI and sudden death compared with the control group. When comparing bezafibrate therapy (400 mg per oral daily) to pravastatin 10 mg per oral daily, RLP-C was reduced more by pravastatin (bezafibrate −16.0% versus pravastatin −40.6%; p<0.05); in contrast, RLP-TG was reduced more by bezafibrate (−55.2 versus −35.0%; p<0.05).80 Bezafibrate significantly suppresses postprandial elevation of RLP-C by approximately 50% relative to placebo in patients with metabolic syndrome.81
In the field trial, 9,795 participants with type 2 diabetes were randomised to receive either fenofibrate or placebo.82 Although the primary composite endpoint for the trial was negative, some individual endpoints, such as nonfatal MI and need for revascularisation, were reduced by 21% (p=0.01) and 24% (p=0.003), respectively. In a post hoc analysis, among participants with high TG (>200 mg/dl) and low HDL-C, the primary composite endpoint was reduced by 27% (p=0.005). Among 20 hypertriglyceridaemic men, fenofibrate reduced RLP-C by 75.4%, and decreased VLDL subclasses 3–6 (larger to smaller) and IDL by 49.3, 39.9, 65.5, 84 and 62.8%, respectively, as measured by nuclear magnetic resonance. Small, dense LDL was reduced by 42%.83
Fenofibrate treatment lowers fasting triglycerides (−46.1%; p<0.0001) and postprandial TG (−45.4%; p<0.0001), with concomitant reductions in postprandial levels of large (−40.8%; p<0.0001) and medium (−49.5%; p<0.0001) VLDL particles when evaluated in 59 men with fasting hypertriglyceridaemia.84 In addition, fenofibrate has been investigated for its potential anti-inflammatory effects. A case–control study investigated the effects of fenofibrate on C-reactive protein (CRP) levels in 280 patients with hypertriglyceridaemia. The study found that fenofibrate significantly reduced CRP levels in patients with a baseline CRP level ≥1 mg/l. This suggests that fenofibrate may have anti-inflammatory properties, specifically in individuals with elevated CRP levels.85
The combination of fibrates and statins has also been studied. The ACCORD trial included 5,518 participants with type 2 diabetes and showed that combining simvastatin with fenofibrate provides no significant benefits in reducing CV risk in the majority of high-risk participants compared with the simvastatin monotherapy group. The annual rate of the primary outcome was 2.2% in the fenofibrate group compared with 2.4% in the placebo group, with a HR of 0.92 (95% CI [0.79–1.08]; p=0.32).86 In another trial, 274 participants with a history of CAD and elevated RLP-C were randomised to either bezafibrate or pravastatin. The impact of these drugs on RLP-C and CV outcomes was measured. After 1-year follow-up, RLP-C levels were reduced more by bezafibrate than pravastatin (37 versus 25% from baseline, respectively), and it was associated with a significant reduction in CV events in bezafibrate-treated participants compared with the pravastatin-treated group (p<0.01).87 The study reported that the reduction of RLP-C levels by 1 SD (1.1 mg/dl) decreased the risk of CVD events by up to 45% (95% CI [33–98]; p=0.03).
Ω-3 Fatty Acids
The Ω-3 FFA have three formulations that are currently Food and Drug Administration-approved in the US: Epa nova, which is a mixture of Ω-3 fatty acids in FFA form; eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid, (Lovaza, Omtryg and generics); and Vascepa, an icosapent ethyl (EPA ethyl esters).88–90 These Ω-3 fatty acids prescriptions have been reported to reduce plasma TG, VLDL-C and non-HDL-C levels in patients with severe hypertriglyceridaemia and to increase HDL-C levels with DHA-containing formulations.91
Multiple clinical trials and meta-analyses have studied the impact of omega-3 FFA and CV outcomes. The Japan EPA Lipid Intervention Study examined the effects of EPA on patients with hyperlipidaemia on a statin background in participants without evidence of pre-existing CAD at baseline. The study reported that EPA therapy suppressed the risk of ASCVD events, with a HR of 0.47 (95% CI [0.23–0.98]; p=0.043) in patients with TGs >200 mg/dl.92 The EVOLVE trial reported that participants who received EPA and DHA had a significant reduction of fasting serum TGs from baseline of up to −30.9% (p<0.001), compared with a −4.3% reduction in the group receiving olive oil.93 The results were associated with a decrease in RLP-C and an increase in LDL-C, which is typical for EPA/DHA combination therapy. The ROMANTIC trial confirmed the significant effect of omega-3 FFA when combined with rosuvastatin in providing incremental reductions in TGs and non-HDL-C compared with rosuvastatin monotherapy.91
The REDUCE-IT reported that participants who received 2 g of icosapent ethyl twice daily (total daily dose, 4 g) experienced a lower incidence of CV death, nonfatal MI, nonfatal stroke, coronary revascularisation or unstable angina compared with the placebo group by 35% (95% CI [0.68–0.83]; p<0.001).94 The trial also demonstrated a 20% reduction in mortality over and above that observed with statin monotherapy. Although EPA monotherapy works well, the combination of EPA/DHA is not efficacious, as shown by the STRENGTH trial.95 The STRENGTH trial demonstrated that among statin-treated patients at high CV risk, the addition of the carboxylic acid forms of EPA and DHA, compared with corn oil, to usual background therapies resulted in no significant difference in a composite outcome of major adverse CV events, with a HR of 0.99 (95% CI [0.90–1.09]; p=0.84).
In a group of patients on dialysis, 1,800 mg of EPA (icosapent ethyl) administered for a period of 3 months reduced remnant lipoproteins by 52%.96 When evaluated in patients with fasting TGs >500 mg/dl, icosapent ethyl reduces RLP-C and ApoC-III by 23 and 16%, respectively.
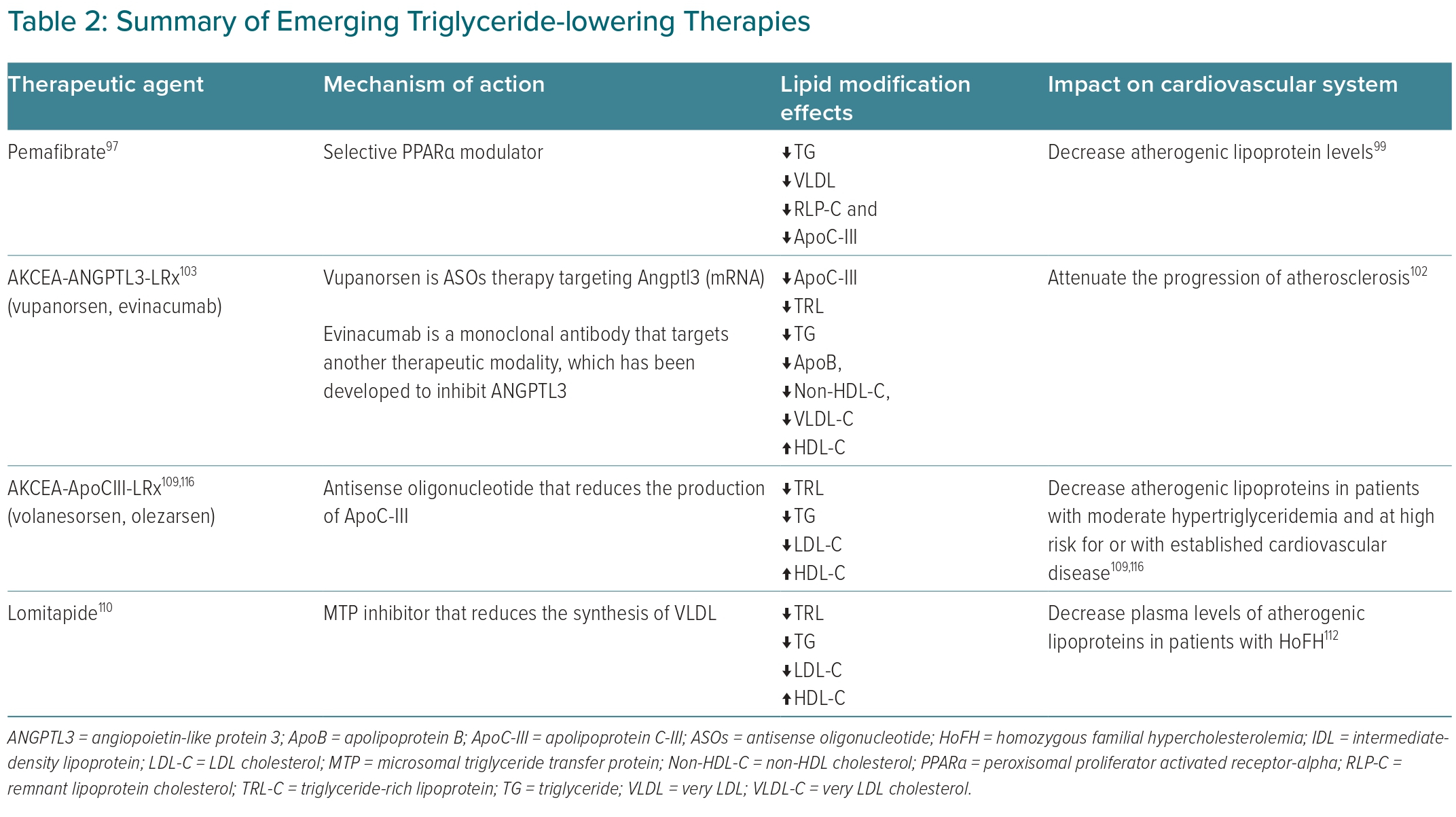
Emerging TG and TG-rich Lipoprotein-lowering Therapies
New therapy targets are emerging, as studies further define and elucidate the mechanism of TRL metabolism and its proatherogenic properties. These agents target the key regulators of TRL metabolism, including LPL modulators and an apoprotein inhibitor (Table 2).
Pemafibrate
Pemafibrate is a novel therapy that acts as a selective peroxisomal proliferator-activated receptor-α modulator.97 Two multicentre, randomised clinical trials were conducted to evaluate the efficacy and safety of pemafibrate to treat residual hypertriglyceridaemia in combination with a statin.98 The studies found a significantly greater reduction in fasting TG levels by approximately 50% in the combination therapy group compared with the statin monotherapy group (p<0.001). Similarly, another clinical trial concluded that pemafibrate decreases TRLs and attenuates the progression of atherosclerosis.99
A recent clinical trial conducted in 68 European sites reported that pemafibrate treatment in combination with a statin reduced TG by up to (−54.4%), with a concordant improvement in other markers for TRL metabolism, including reductions in ApoB48 (−63.4%), ApoC-III (−36%) and non-HDL-C (−9.1%) seen in the pemafibrate 0.2 mg once daily group, and an increase in HDL-C levels by up to 12.9%.100 Although multiple studies confirmed the favourable effect of pemafibrate on the lipid/lipoprotein profile, a recent multinational clinical trial (PROMINENT) failed to show a significant reduction in CV events compared with the control group, with a HR of 1.03 (95% CI [0.91–1.15]; p=0.67).101 Thus, further studies are needed to assess the effectiveness of pemafibrate in reducing CV events.
The failure of the PROMINENT trial can be attributed to several factors. One of the key observations was the reduction in TG (26.2%) and RLP-C (25.6%). These changes were a result of increased LPL activity coupled with a decrease in ApoC-III levels. These changes were counterbalanced by lack of change in non-HDL-C (−0.2%), and the elevations in ApoB (4.8%) and LDL-C (12.3%) relative to placebo. It is unlikely that the increase in RLP-C clearance is attributable to an upregulation of RLP receptors on hepatocytes, as ApoB and non-HDL-C did not decrease. The more plausible explanation is that, as pemafibrate activated lipoprotein lipase, the increase in lipolysis led to a decrease in remnant lipoproteins because of their increased conversion to LDL. The increase in VLDL conversion and the rise in LDL-C were not accompanied by an upregulation in LDL receptor expression. The use of pemafibrate constitutes a suboptimal approach to demonstrate the impact of RLP reduction on CVD, as total atherogenic lipoprotein burden in serum did not change (i.e. ApoB and LDL-C increased, non-HDL-C was neutral). Hence, it is not entirely surprising that the trial was negative.
Therapies Targeting Angiopoietin-like Protein 3
AKCEA-ANGPTL3-LRx is an investigational antisense oligonucleotide (ASO) therapy targeting ANGPTL3 messenger RNA (mRNA).102 Epidemiological and genome-wide association studies have linked loss-of-function variants in ANGPTL3 with lower levels of plasma lipoproteins. A Phase I randomised, double-blind, placebo-controlled clinical trial in healthy adults was conducted to assess the efficacy and safety of ANGPTL3-LRx.102 This trial reported that after 6 weeks of ANGPTL3-LRx therapy, participants in the multiple-dose groups had reductions in levels of ANGPTL3 protein by −46.6 to −84.5%, associated with reductions in levels of TG (−33.2 to −63.1%), LDL-C (−1.3 to −32.9%), VLDL (−27.9 to −60.0%), non-HDL-C (−10.0 to −36.6%), ApoB (−3.4 to −25.7%) and ApoC-III (−18.9 to −58.8%). Treatment of Western-diet-fed LDL receptor knockout mice with ANGPTL3 ASO attenuated the progression of atherosclerosis by 52% (p=0.002) compared with the control group in a dose-dependent pattern. Vupanorsen is an N-acetyl galactosamine-conjugated ASO that selectively inhibits ANGPTL3 protein synthesis.103 The N-acetyl-galactosamine group allows vupanorsen to bind to N-acetyl-galactosamine receptors on the hepatocyte surface, thereby increasing tissue-specific penetration. Vupanorsen treatment provides significant reductions in TGs (−47%) and ANGPTL3 (−56%) compared with placebo. Vupanorsen improves the lipid/lipoprotein profile by reducing ApoC-III (−58%), RLP-C (−38%), TC (−19%), non-HDL-C (−18%) and ApoB (−9%). There were no serious adverse events reported secondary to vupanorsen treatment.103
Evinacumab is another therapeutic modality that inhibits ANGPTL3.34 It is a monoclonal antibody that reduces LDL-C and TGs in homozygous familial hypercholesterolaemia. Findings from two Phase I clinical trials conducted in participants with hypertriglyceridaemia have reported a dose-dependent reduction in TGs observed in both studies, with a maximum decrease in TGs by up to −83.1% in the treatment group compared with the placebo group. Evinacumab reduces RLP-C by up to 50%.104
Therapies Targeting Apolipoprotein C-III
Volanesorsen and olezarsen are ASOs that regulate TRL metabolism by targeting ApoC-III production, and effectively decrease TRL and TG levels in patients with hyperchylomicronaemia.105 A preclinical Phase I/II double-blind, placebo-controlled, dose-escalation clinical trial was conducted in participants without an established ASCVD history and elevated TGs levels to assess the efficacy of volanesorsen.106 The study reported that the single-dose treatment group had a reduction of ApoC-III by up to 92% and a reduction of TGs by up to 77%. The multiple-dose treatment group showed a similar pattern and reduced ApoC-III by up to 89% and TG by up to 66% in a dose-dependent manner. Additionally, a significant reduction in total cholesterol, ApoB, non-HDL-C, VLDL-C and increases in HDL-C were concomitantly observed.
Volanesorsen was approved in May 2019 in Europe for managing familial hyperchylomicronaemia syndrome based on the promising results from several multinational clinical trials, including the Phase III APPROACH and COMPASS studies.105 The APPROACH trial reported that volanesorsen decreased TGs levels to <750 (mg/dl) in 77% of patients with familial chylomicronaemia syndrome.107 During the study, thrombocytopenia and injection-site reactions were reported as adverse events.107 Data from the COMPASS trial was consistent with the previous clinical trial findings and showed that volanesorsen reduced mean plasma TG levels by −71.2% (p<0.0001) from baseline to 3 months compared with the placebo group.108 The side effects reported during the study were mainly secondary to injection site reactions. Volanesorsen is not approved in the US due to its capacity to induce thrombocytopenia.
Olezarsen is another novel N-acetyl-galactosamine-conjugated ASO targeted to hepatic ApoC-III mRNA to inhibit ApoC-III production.109 A randomised, dose-ranging study was conducted to assess olezarsen efficacy, safety and tolerability, and showed that fasting TGs levels decreased by up to 60% in the treatment group compared with the control group. Olezarsen concomitantly reduced ApoC-III, VLDL, non-HDL-C and ApoB. Finally, The most reported common adverse event documented was mild erythema at the injection site. Olezarsen therapy is not associated with thrombocytopenia.
Lomitapide (Juxtapid) inhibits microsomal triglyceride transfer protein, which is the enzyme essential for VLDL assembly and secretion.110 Lomitapide decreases TRLs and TGs levels. A single-arm, open-label, Phase OOO study of lomitapide that included participants with homozygous familial hypercholesterolaemia resulted in lower LDL-C, TGs, lipoprotein (a) and ApoB levels compared with the control group.111
Similarly, a Phase III dose-escalation and safety study of lomitapide conducted in Japanese participants with homozygous familial hypercholesterolaemia reported a reduction of the levels of TGs by 54.8% and non-HDL-C by 37.4%.112 Participants continued the study with acceptable safety and tolerability, and without serious side-effects or death reported. However, despite the lipid/lipoprotein profile’s favourable outcomes, the safety profile of lomitapide has shown some concerns; hepatic transaminitis and fatty liver were the most serious adverse events reported in the treatment group compared with the control group.110
Conclusion
It is established that LDL-C is the essential causal lipoprotein in developing atherosclerosis. Thus, therapies targeting LDL-C are still the principal strategy for the primary and secondary prevention of ASCVD. However, patients continue to develop ASCVD events despite a significant reduction of LDL-C, which reflects residual risk arising from other ASCVD risk factors. The findings from observational and genetic epidemiological studies suggest a causal role for TRLs and their remnants as potentially independent risk factors for ASCVD. However, the differences in separation techniques and the lack of standardisation for measuring TRLs and RLPs limit our ability to infer strength of association of specific lipoprotein fractions. Thus, to minimise inconsistent findings and controversy over the role and mechanism of TRLs in CVD, standardised strategies are needed to better understand the nature of the relationship between ASCVD risk and TRLs. Finally, the results of ongoing clinical trials testing the impact of TRLs and RLP-C on ASCVD will help better define whether and how these lipoprotein species with their TGs and cholesterol should be therapeutically targeted.