










In this article, we discuss severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the role of the renin–angiotensin–aldosterone system (RAAS) in the pathogenesis of coronavirus disease 2019 (COVID-19), with special emphasis on the association between gene variants and the risk of infection and disease severity.
SARS-CoV-2 was first identified in Wuhan, China in December 2019. Due to the similarity of its genetic sequence and pathophysiological mechanisms when compared with the coronavirus that caused severe acquired respiratory syndrome (SARS-CoV-), this novel coronavirus was designated SARS-CoV-2. The virus has rapidly spread worldwide with millions of people affected and a significant number of deaths.
The main manifestation of COVID-19 is a respiratory disease that varies from almost no symptoms to a pneumonia that requires hospitalisation. Some patients will have severe pneumonia that needs to be treated with mechanical respiratory support in an intensive care unit (ICU). The rate of mortality was not clear because of the heterogeneous values between countries. It was only clear that the mortality was higher among older patients.
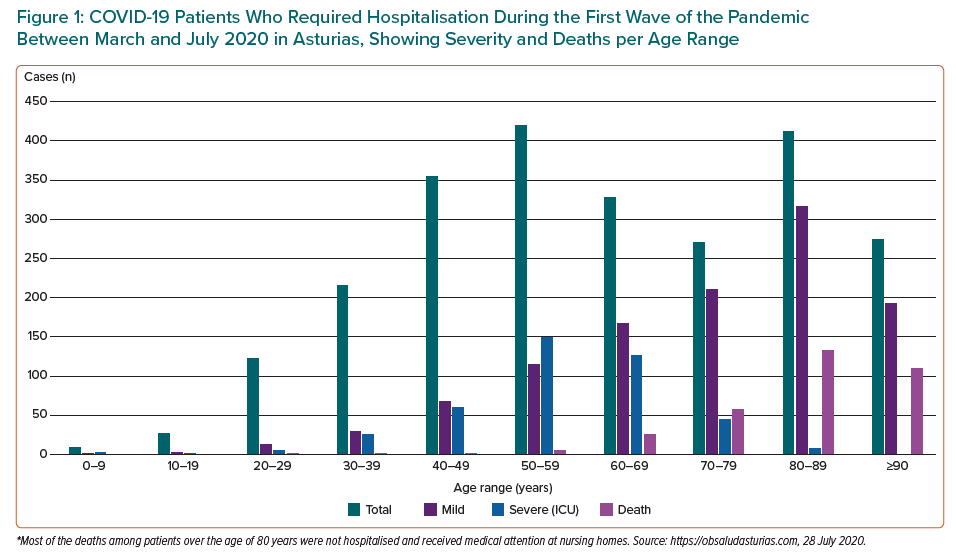
Figure 1 shows the age distribution of people with COVID-19 who required hospitalisation (either non-severe or severe ICU cases during the first wave of the pandemic from March to July 2020) in Asturias in northern Spain, which has a total population of about 1 million. On 28 July 2020, there were a total of 2,433 confirmed cases in Asturias, of whom 42% were men. Of these 2,433 cases, 1,117 were hospitalised and 421 needed respiratory support in the ICU. More men had severe cases and needed to be treated in an ICU. There was a total of 334 COVID-19 deaths in the first wave of the pandemic (March–July 2020).
COVID-19 is suspected by its clinical symptoms and is confirmed by a positive genetic test based on the amplification of the viral genome from nasopharyngeal swabs or bronchoalveolar lavage. Despite COVID-19 presenting a challenge to health systems because of the large number of patients who require hospitalisation, most people who test positive for the virus show none or very mild flu-like symptoms.
RAAS and COVID-19 Infection
Many of the disease-causing mechanisms of SARS-CoV-2 have been elucidated. Its genome has been well characterised, and this facilitates the viral detection and the identification of protein epitopes that can be used in vaccine design. Similar to SARS-CoV-1, this coronavirus binds to the human angiotensin-converting enzyme 2 (ACE2) protein in cell surfaces. ACE2 is a high-affinity receptor for the viral spike (S) protein. After binding to ACE2, a membrane protease (TMPRSS2) modifies the S-protein facilitating the fusion of the cell membrane with the viral membrane. Once in the cytoplasm, the viral genome dictates the synthesis of proteins necessary for the replication and spread of SARS-CoV-2.
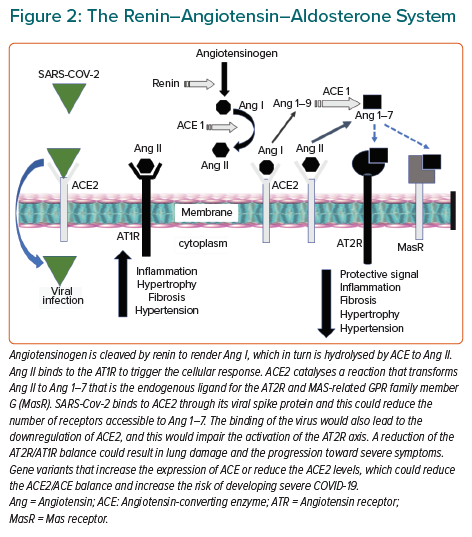
The cellular levels of ACE2 depend on several factors, including the activity of other RAAS components, gene polymorphisms and clinical conditions, such as hypertension (Figure 2). All these factors could contribute to a person’s risk of infection and disease severity. The role of ACE2 in SARS-CoV-2 infection raised some initial concerns about the effect of ACE inhibitors (ACEIs) and angiotensin-2 (Ang II) receptor blockers (ARBs) on this disease.
COVID-19, RAAS and Cardiovascular Disease
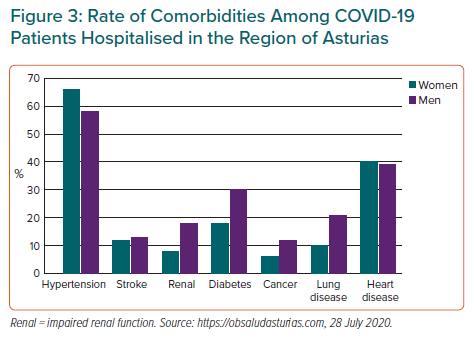
Initial data showed that cardiac injury may be present in up to 30% of patients who had been hospitalised with COVID-19.1–3 Evidence clearly shows that in addition to older age, hypertension, diabetes and cardiovascular disease (CVD) were risk factors for developing severe COVID-19, and patients with cardiovascular comorbidities were at a higher risk of death from COVID-19 (Figure 3).
The putative mechanisms of myocardial injury in COVID-19 include ischaemia due to circulatory and respiratory failure, epicardial or intramyocardial small coronary artery thrombotic obstruction due to increased coagulability, and myocarditis secondary to systemic inflammation or direct binding of the virus to ACE2 in cardiomyocytes. Persistent immune activation upon SARS-CoV-2 infection would also increase the risk of developing dilated cardiomyopathy.4
ACE2 is a membrane-bound carboxy-dipeptidase expressed in many cell types and tissues, including the upper airway, lung, heart, kidney and small intestine.5 The wide distribution of ACE2 would make these organs accessible for viral infection and could explain some of the extrapulmonary manifestations of COVID-19 and the SARS-CoV-2 genome has been amplified from samples obtained from several tissues. Being male or older are the main predictors of severe COVID-19. Age and sex differences for disease progression between people with COVID-19 could be explained in part by the fact that the rate of ACE2 expression varies with age and seems to differ between men than women.6
The physiological effects of ACE and ACE2 are mediated by two angiotensin II receptors, type 1 (AT1R) and type 2 (AT2R). Each display opposite responses, with ATR1 mediating vasoconstriction, proliferation, fibrosis and inflammation, and AT2R mediating vasodilation, antifibrosis and anti-inflammation responses (Figure 2). The two converting enzymes are thus seen as positive and negative regulators of the RAAS response and the deregulation of the ACE/ACE2 balance could result in cardiovascular disease, hypertension, cardiac hypertrophy and heart failure, among other conditions. ACE2 has been shown to be an essential regulator of cardiac function in a study that showed ACE2 knockout mice developing severe left ventricular dysfunction, and the reduction in cardiac contractility being restored by a reversion of the knockout phenotype. In addition, a DNA variant in the human ACE2 gene has been associated with the risk of cardiovascular death.7,8 This could have implications for COVID-19 in terms of a genetic susceptibility to infection or the risk of developing a severe form of the disease.
The fact that pulmonary lesions induced by the SARS coronavirus were more aggressive in ACE2 knockout mice compared with wild-type mice (an effect that was attenuated by blocking the RAAS) suggests that ACE2 expression levels could also influence the severity of respiratory disease found in patients with COVID-19.9
SARS-CoV-2 Infection and Cardiac Injury
Cardiac cells express ACE2 which means that myocardial infection by SARS-CoV-2 could be possible. The viral genome has been amplified in post-mortem myocardial tissue from patients who died from SARS, but the notion that there is a direct effect of SARS-CoV-2 on myocarditis is controversial. The systemic inflammation and metabolic imbalance could also explain the cardiac arrhythmias seen in some patients. COVID-19 patients also showed significantly elevated levels of D-dimer, a fibrin degradation product of thrombosis.10 The elevation of this marker seems to be a strong predictor of mortality from COVID-19, although the prevalence of acute coronary syndromes due to increased thrombotic activity is unknown.
Since COVID-19 has arisen from a novel virus, we do not have data about its long-term manifestations. It could be, for example, that the inflammatory exacerbation could enhance the development of atherosclerotic plaques and increase the risk for future ischaemic episodes. Several studies have shown that ACE2 is implicated in atherosclerosis pathophysiology. Among others, ACE2-deficient mice showed increased levels of proatherogenic mediators with an impaired endothelium-dependent relaxation that was attenuated by the blockade of angiotensin 1–7; while the overexpression of ACE2 in human endothelial cells stimulated endothelial cell migration and limited the expression of monocyte and cellular adhesion molecules.11,12 Moreover, the inhibition of ACE2 in atherosclerosis-prone apolipoprotein E-deficient mice increased the proatherogenic effect of a high-fat diet.13
Some authors have speculated that the binding of the virus to the receptor could reduce the amount of ACE2 accessible for Ang II, with a downregulation of the AT2R-mediated response (Figure 2). This hypothesis is plausible considering the reported downregulation of ACE2 among mice that had been infected with SARS-CoV-2.9 Moreover, injecting the spike protein into mice downregulated ACE2 in the lung and worsened acute lung failure, which was attenuated by blocking the renin-angiotensin pathway.9 Another study has shown significantly higher levels of ACE2 in the lungs of patients with comorbidities associated with severe COVID-19 compared with controls.14 This finding might explain the higher risk of developing severe COVID-19 in people with such comorbidities, but there is no evidence that SARS-CoV-2 has a significant effect on ACE2 expression and activity.
The involvement of ACE2 and its deregulation in COVID-19 patients could increase the risk of future acute coronary events, particularly in people who are already predisposed to these events, such as those with diabetes, hypercholesterolaemia or hypertension. Although this hypothesis is plausible based on current evidence, it requires experimental validation. Due to the large number of COVID-19 cases, a new line of research to define the long-term cardiovascular effects is recommended.
RAAS Drugs and COVID-19: A Cause for Concern?
During the early weeks of the pandemic there were concerns about the effect that antihypertensive drugs that target the RAAS may have on the risk of COVID-19 infection and disease outcomes.15–18 ACE inhibitors and AT1R blockers have a protective cardiovascular effect by limiting the AT1R-mediated response, but also increase the ACE2 expression enhancing the vasoprotective axis of the RAAS. Higher levels of ACE2 would also reduce the risk of developing atherosclerosis. It is not so surprising that the identification of ACE2 as the receptor for SARS-CoV-2 raised speculations about the detrimental effect of RAAS-drugs in relation to COVID-19. ACEIs and ARBs promote the expression of ACE2 perhaps making the patients treated with these drugs more susceptible to infection, which is important when we consider the relationship between COVID-19, hypertension and mortality.
At least one study has addressed the association between ACEIs and ARBs and testing positive for COVID-19. In this retrospective cohort of 18,472 patients, the authors found no association between these drugs and a positive COVID-19 test.15 The most recent large, well-conducted observational studies also suggest no harm from these drugs. Among others, a study including 1,128 adult patients with hypertension diagnosed with COVID-19 found a lower mortality rate among patients treated with ACEI/ARB after adjustment for age, sex, comorbidities and in-hospital medications. Mortality was also lower in the ACEI/ARB group.16 A large population-based study performed in Italy examined 6,272 COVID-19 patients and population controls. The use of ACEIs and ARBs was more common among those with COVID-19 because there was a higher prevalence of the disease among people with CVD.17 The use of these drugs did not show any association with disease outcomes, severity or death. There was also no association between sex and the clinical variables observed.
A simple picture of the effect of ACEIs and ARBs would show the net balance between the enhancement of ACE2 levels that might be detrimental due to the increased amount of receptor accessible for viral infection, but also beneficial by promoting the AT2R-mediated response (Figure 2). Although SARS-CoV-2 might downregulate ACE2, reducing its protective effects and exacerbating injurious Ang II effects, retrospective observational studies do not show that ACEI or ARB users are at increased risk of infection or severe symptoms, and medication changes should be considered based on the individual patient’s clinical condition.18,19
Genetics of RAAS in Cardiovascular Disease and COVID-19
One of the most intriguing issues that the COVID-19 pandemic has raised is the heterogeneous manifestations between individuals exposed to SARS-CoV-2. Many people who test positive for the virus show very mild or even no symptoms and would not require medical attention. The identification of these asymptomatic carriers is a major challenge because they can transmit the virus to others and spread the disease. In other cases, the new coronavirus triggers a deleterious response that could require hospitalisation and admission to ICU. As indicated above there are several well-recognised risk factors for COVID-19 severity and mortality, such as sex, that could be associated with either biological differences between men and women or a higher frequency of smoking and other acquired respiratory risk factors among men.20,21
It has been proposed that the levels of several cellular components required for the viral infection depends on the patient’s sex. For instance, ACE2 expression seems to be higher in women than men and it decreases with age. A study that measured plasma ACE2 concentrations in older people with heart failure found higher levels in men than in women and use of neither an ACEI nor ARB was associated with higher plasma ACE2 concentrations.6 A complete vision of the pathogenesis of the effects of SARS-CoV-2 must consider non-modifiable factors, such as age and sex, but also genetic variability in each individual.22 In this context we can use our extensive knowledge of the genetics of the RAAS and how the variation of the genes that encode proteins in this pathway modulate the risk of developing hypertensive and cardiovascular traits.
A common insertion/deletion polymorphism in the ACE gene (ACE-I/D) is one of the best characterised human variants and has been extensively studied in several traits. The deletion allele is associated with higher expression of ACE and deletion homozygotes have significantly higher ACE blood levels compared to insertion carriers. ACE-D has thus been associated with diseases in which the RAAS could be overactive with increased circulation levels of Ang II, such as hypertension, cardiac hypertrophy, heart failure or nephropathies.23,24 This polymorphism has also been associated with the response to physical training because the insertion seems to be more prevalent among elite athletes. There are several mechanisms by which ACE activity could regulate the progression of COVID-19, including the modulation of the ACE2 expression that would make some individuals more susceptible to COVID-19 than others. Some authors have speculated that ethnic differences in ACE-I/D frequency could explain the apparently heterogeneous inter-population rate of infection. The expression of the two ACEs seems to correlate and might be influenced by hypertension status.25 An interesting hypothesis is that functional variants in the two ACEs (and other RAAS genes) could modify the ACE/ACE2 balance and disease susceptibility and severity. The association between ACE polymorphisms and acute respiratory failure is controversial.26–28 Whether these variants define COVID-19 severity and mortality should be investigated.
The ACE2 Gene: A Putative Player in COVID-19
The ACE2 gene is on the X chromosome, so men carry only one copy, a fact that could help explain why the disease seems to be more severe among men. Because women carry two gene copies, a functional variant that confers disease risk when homozygosis would be less frequent among women. The role of ACE2 in cardiovascular and lung disease could explain not only any genetic association with COVID-19 outcomes, but also the susceptibility to SARS-CoV-2 infection. The lack of the C-C chemokine receptor type 5 (CCR5) due to a common mutation confers resistance to HIV infection, and individuals with a complete lack of CCR5 do not develop AIDS in spite of exposure to HIV. A similar mechanism could happen for ACE2 in cases resistant to infection by SARS-CoV-2, but this is unlikely because contrary to the viability of individuals with a complete deletion of CCR5, no mutation that abrogates the ACE2 has been reported.
In fact, the data from human genome sequences reveals that the ACE2 gene shows a very low polymorphic rate in the coding sequence, suggesting a constraint against mutations. Our group has sequenced the ACE2 coding sequence in 50 patients with COVID-19, including 25 severe cases, and no variant affecting the protein sequence was identified.29 Of course, ACE2 variants related with reduced protein expression could make the cells less prone to infection and could result in partial resistance among carriers, but at the same time, this would reduce the beneficial effect of ACE2 expression making the individuals more susceptible to adverse COVID-19 outcomes.
Conclusion
The RAAS is implicated in the COVID-19 pandemic through the pivotal role of ACE2 in viral infection. Sex- and age-dependent ACE2 expression could partly explain the higher risk of severe COVID-19 and mortality among older men. In addition, the imbalance of the components of the RAAS could explain the frequency of hypertension and other cardiovascular traits among severe cases. The current evidence suggests that ACEIs or ARBs do not increase the risk of infection or the development of severe COVID-19. A genetic predisposition due to ACE2 gene variants could contribute to define the susceptibility and outcomes of COVID-19. However, this is mere speculation in most COVID-19 literature and although it is based on a plausible hypothesis there is a lack of experimental validation. We are currently viewing the immediate acute effects of COVID-19 in people with cardiovascular comorbidities, but the long-term effects on the cardiovascular system requires the follow-up of patients who have recovered from COVID-19.