










Environmental stressors, such as noise, air or heavy metal pollution, represent important risk factors for the development and progression of cardiovascular disease (CVD).1–5 The WHO estimates that at least 1.6 million healthy life years are lost every year in western European countries alone due to the exposure to environmental noise. Additionally, within the last years, the GBD study established that ambient outdoor air pollution due to particulate matter <2.5 μm (PM2.5) was the fifth-ranking global risk factor in 2015, causing 4.2 million deaths annually, with cardiovascular deaths accounting for most of these deaths.6 Importantly, this statistic does not include other occupational and environmental stressors, such as smoking, household air pollution, mental stress or noise, which, taken together, arguably could outrank the global impact of many classical risk factors in combination. In addition, mercury, lead (Pb) or cadmium (Cd) are toxic elements commonly found in the environment, contributing to hypertension.7 According to several reports, high concentrations of heavy metals in different natural systems, including atmosphere, pedosphere, hydrosphere and biosphere, have become a global issue.8 Importantly, these heavy metals have been and are still commonly used in various areas, especially manufacturing and agriculture.9 Alterations in vascular function were the earliest pathophysiological mechanism described in response to noise, air, or heavy metal (e.g. mercury, Pb or Cd) exposure in humans, and indeed disturbances in endothelial function are a critical initiating event that is widely relevant to almost all classical risk factors.10–14 In the present review, we focus on the evidence supporting the impact of different pollutants and, particularly, its molecular mechanism involved in the development of endothelial dysfunction. Additionally, we focus on to what extent alterations in vascular function are responsible for mediating systemic diseases in response to noise, air and heavy metal pollution. Finally, we examine the epidemiological/observational evidence on the association of environmental risk factors and surrogate markers of endothelial dysfunction.
Endothelial Dysfunction is Triggered by the Environment
Endothelial Function
There are three distinct layers in the wall of an artery (Figure 1A). Firstly, the adventitia has primarily a mechanical role to fasten the vessel to the surrounding tissue and driving the inflammation.15,16 Then comes the tunica media layer that regulates the internal diameter of the vessel. In addition, the smooth muscle cells in this layer possess receptors for neurotransmitters and hormones, which allow direct effects on the vasoconstriction of vessels by stress response systems.15,17,18 Finally, the tunica intima has a specialised monolayer of epithelial cells called the endothelium.15,19,20 The endothelium, which is a significant source of nitric oxide (NO) that is produced by nitric oxide synthases (NOS), and its principal function is to control over vasoconstriction and vasodilation.21–23 A healthy endothelium maintains communication with the media through signalling molecules, of which NO plays a prominent role.24 Endothelial NOS (eNOS) uses the amino acid L-arginine as a substrate to generate NO. NO is primarily involved in vasodilation of vascular smooth muscle cells and prevention of platelet/immune cell adhesion to the vascular wall when it is released into the circulation (Figure 1B). eNOS consists of different domains; the carboxy-terminal reductase domain is homologous to the cytochrome P450 reductase and contains a binding site for the nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD) and calmodulin (CaM).25 Another principal domain is the amino-terminal oxygenase that contains a heme prosthetic group, binding sites for the substrate L-arginine and the cofactor tetrahydrobiopterin (BH4), which is required for the efficient generation of NO by controlling the redox state of the oxy-iron intermediate.26 In the process of constitutive formation of NO, calmodulin activity is modulated by the increase of intracellular Ca2+ levels, thereby enhancing the flow of electrons from NADPH, which represents the signal for activating the reductase function of the enzyme.27,28 The whole regulation of eNOS is shown in Figure 1C. In healthy endothelial cells, eNOS can be activated either by mechanoreceptors that are sensitive to the shear stress generated by blood flow or by acetylcholine (ACh) receptors, all of which leads to altered phosphorylation state of eNOS.28 NO produced by eNOS diffuses to smooth muscle cells and mediates relaxation through cGMP signalling mechanism, finally decreasing the intracellular calcium concentrations in smooth muscle cells and thereby decreasing the contractility of the actinin-myosin-filaments.29 In order to stimulate vascular smooth muscle relaxation, an endothelium-independent approach can be used, where NO is supplied by donor drugs, such as nitroglycerin (GTN). GTN undergoes bioactivation via mitochondrial aldehyde dehydrogenase (ALDH-2) to generate NO, which can directly stimulate vascular smooth muscle relaxation.30
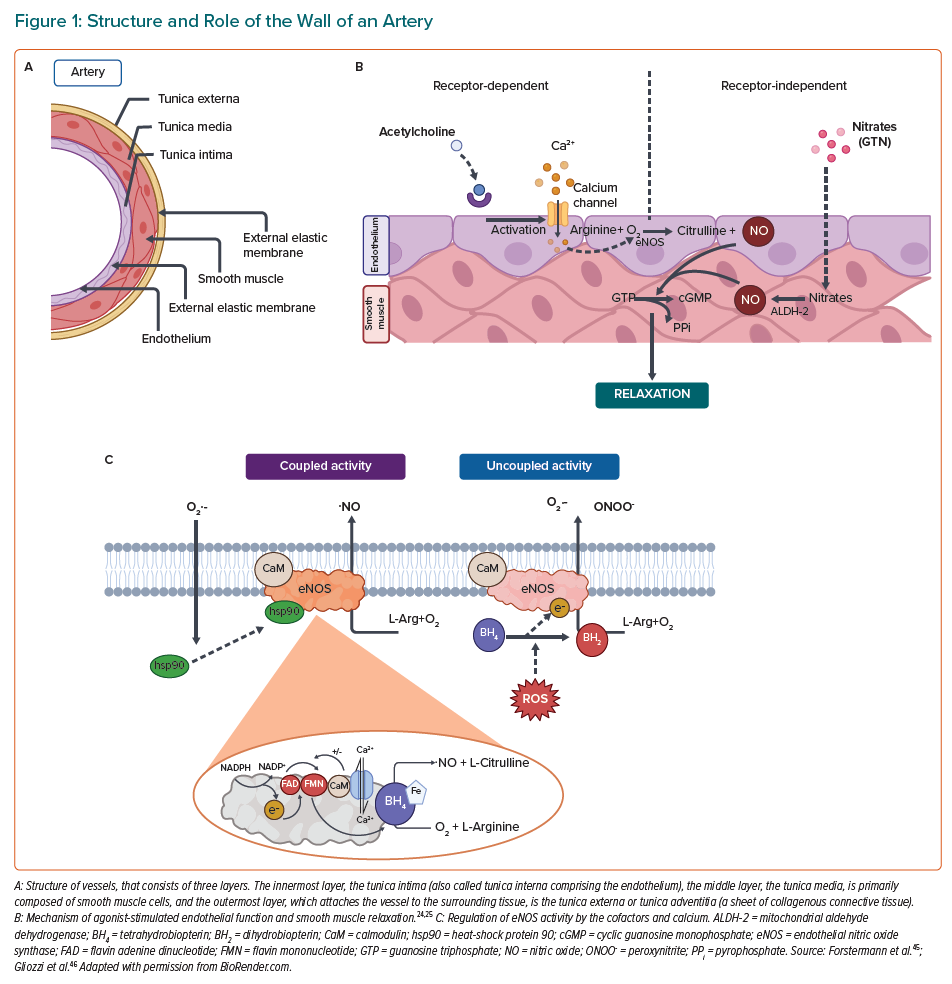
Endothelial Dysfunction Is Induced by Environmental Stress
The inability of a vessel to dilate fully, which arises from a decrease in NO bioavailability caused by reduced production, scavenging by reactive oxygen species (ROS), or both, is known as endothelial dysfunction.24 During the last decades, it has been shown that novel risk factors in the physical environment may facilitate the development of cardiovascular complications such as endothelial dysfunction.31,32 For this reason, endothelial dysfunction in the setting of cardiovascular risk factors, such as hypercholesterolemia, diabetes, chronic smoking or hypertension, is dependent on the production of ROS and the subsequent decrease in vascular bioavailability of NO.14 Additionally, it is known that some stress conditions, such as air or noise pollution, in the setting of cardiovascular diseases (CVD) are characterised by impaired NO signalling and associated with increased circulating stress hormones, cytokines, vasoconstrictors, such as angiotensin II (AngII) and circulating or infiltrating immune cells.33 NO can be scavenged by other free radicals such as superoxide, which ultimately leads to endothelial dysfunction (Figure 1C). When there is a reduction in the bioavailability of BH4, a cofactor of eNOS, because the rate at which it is recycled is slower than the rate of oxidative degradation, it effectively causes eNOS to produce superoxide, rather than NO.26 Reductions in the amount of NO impairs vascular signalling and results in endothelial dysfunction. Bioavailability of BH4 can be altered by the redox status of the cell (i.e. oxidative degradation to BH2) or, since it is endogenously produced, the pathway for BH4 production can be altered. Furthermore, redox modifications to eNOS itself can cause uncoupling, which again leads to superoxide production.26 The disruption of NO synthesis could be affected by several factors, which have a direct correlation with oxidative stress.34
There is evidence that preserving an intact endothelium is a critical component of healthy cardiovascular physiology, since endothelial dysfunction can be detected as an early sign of atherosclerosis and its associated pathologies, hypertension, diabetes and dyslipidemia.31,35–38 Endothelial dysfunction is also present in people with other risk factors for cardiovascular disease, such as in smokers, people with disturbed sleep, chronic inflammation and chronic stress.39–44
Mechanistic Insight into Endothelial Dysfunction Consequences of Noise Exposure and Its Outcomes
Adverse Cardiovascular Effects of Noise in Animals
The association between noise exposure, endothelial dysfunction and cardiac damage has been suspected for several decades, and investigation into the mechanism by which damage occurs is well underway. An early work in the field was a study where it was demonstrated that cardiac fibrosis and ischaemic myocardial lesions were present in the hearts of rats exposed to relatively low levels of noise (65 dB(A)) over the course of 52 weeks. Herrmann et al. demonstrated that noise stress may also cause a change in microvessel anatomy. Spontaneously hypertensive rats (SHR) were found to have more microvessels with an outer diameter of >19 µm and more severe cardiac fibrosis and extensive ischaemic myocardial lesions, but these effects were not seen in normotensive rats.47 Moreover, in 2013, fibrosis was also observed following noise exposure at 90 dB.48 Atrial fibrosis was reported in conjunction with reductions in connexin 43 levels following 120 dB noise.34 Signs of necrosis and inflammation were also reported in the myocardium of rats exposed to moderate noise levels.49 In terms of impact on the vessels, lumen narrowing was reported in the cochlear vessels following noise exposure in guinea pigs, which normalised several days after noise cessation.50 This report was later confirmed and alleviated in guinea pigs with treatment with etanercept, a TNF inhibitor.51 Other studies established that white noise exposure of rats for periods of 2–8 weeks (85–100 dB(A)) significantly impaired endothelium-dependent vasodilation (measured by acetylcholine-dependent relaxation in thoracic aorta or mesenteric arterial rings), increased the sensitivity to the vasoconstrictor serotonin, decreased the lumen sizes of microvessels, increased systolic blood pressure by 25–37 mm Hg and increased circulating markers of oxidative stress.52–55
Noise has been associated with annoyance, stress, sleep disturbance and impaired cognitive performance.56 In some animal noise models, data was generated that may explain the mechanism of aircraft noise-induced vascular dysfunction, oxidative stress and inflammation.57,58 Elevated levels of noradrenaline, adrenaline, angiotensin II (Ang II) and, subsequently, cortisol were present after an overactivation of the sympathetic nervous system. Next, Ang II activates endothelial NADPH oxidase via activation of protein kinase C (PKC), causing oxidative stress, which may induce direct scavenging of NO and eNOS uncoupling through oxidation of BH4 and eNOS S-glutathionylation. ROS play a key role in linking different pathways, including PI3K/Akt signalling, the FOXO transcription factors, TGF-β1 and NF-kB signalling, as well as the endothelin-1 (ET-1) system, increasing the circulating levels of interleukin (IL)-6 and the expression of vascular adhesion molecules. Superoxide and nitric oxide produced by infiltrating immune cells (neutrophils, NK cells and monocytes/macrophages) promote the formation of peroxynitrite and subsequent oxidative damage in the form of 3-nitrotyrosine (3-NT), malondialdehyde (MDA) and 4-hydroxynonenal-(4-HNE) positive proteins and other cellular oxidative damage. The uncoupling of eNOS not only reduces NO production but also potentiates the pre-existing oxidative stress. Endothelial NO production is further reduced by glucocorticoids like cortisol, leading to impaired vasodilation and increased blood pressure. The overproduction of catecholamines (noradrenaline (NA), adrenaline (A)) and ET-1 enhances contraction, which is further potentiated by glucocorticoids. In terms of oxidative stress, in mice (C57BL/6J), aircraft noise induces Nox2 expression and uncoupling of the eNOS, leading to cardiac superoxide formation. Superoxide has been well established as a nitric oxide scavenger resulting in a peroxynitrite production and, correspondingly, a progressive reduction in plasma NO2- was found in our last hypertension study.57–60 In addition, oxidative stress affects the health of the endothelium; this event also affects blood pressure due to the importance of the endothelium as a regulator of vascular tone. In our recent studies, gp91phox deletion and LysM+ cells ablation appear to be effective against developing hypertension from noise exposure.58,61 All these vascular alterations support the development of metabolic disorders as envisaged by increased blood glucose levels, insulin resistance, obesity, hypertension and atherosclerosis in noise-exposed mice.57,60,62 Upon chronic activation of stress response pathways, cardiovascular risk factors can emerge or be exacerbated and lead to cardiovascular or neuronal diseases.63–65 In addition, it was shown that also circadian dysregulation plays a key role in cardiovascular disorders caused by noise exposure.66 More recently, in 2022, it was demonstrated that endothelial dysfunction and inflammation were present during the entire 28 days of aircraft noise exposure (at a mean sound pressure level of 72 dB (A) and peak levels of 85 dB (A)).67 Altogether, these studies provide a solid foundation, given the apparent relationship between noise exposure, endothelial dysfunction and the subsequent vascular and cardiac damage. Moreover, all of the mentioned ones further illustrate health side effects of short and long-term noise exposure and further strengthen a consequent implementation of the WHO noise guidelines in order to prevent the development of noise-related future cardiovascular disease. For a selected overview of the mentioned experimental noise exposure protocols in the context of endothelial dysfunction, see Supplementary Material Table 1, whereas noise exposures for the purposes of studying other pathologies (e.g. acoustic damage) are not included.
Negative Consequences on Endothelial Function Caused by Air Pollution in Animals
Air Pollution
Air pollution is an overarching term used to describe, gaseous, liquid and solid pollutants that are present in the air, and that can arise from both human activity (industry, agriculture, farming, etc.) and natural sources (volcanic dust, soil dust, wild fires, etc).68 Historically, the main focus of the air pollution research field included the gaseous pollutants like nitrogen oxides (NOx), carbon monoxide (CO), sulphur dioxide (SO2) and ozone (O3), since they are common side products of large industrial processes.11 Other gases, such as volatile organic compounds (VOCs), non-methane hydrocarbons and polycyclic aromatic hydrocarbons (PAH), have recently received proper scientific attention. Solid and liquid components of air pollution, termed particulate matter (PM), have recently gained attention from researchers and legislators, as the detrimental effects of PM on human health have been recognised in a growing number of studies.2 Ambient (outdoor) PM was ranked seventh among the risk factors for attributable disability-adjusted life years (DALYs) in the 2019 Lancet’s GBD study, up from 13 place in 1990, but household air pollution fell from fourth to tenth place, showing the importance of this stressor.69 The GBD study also estimated that 6.5 million yearly deaths globally can be attributed to air pollution, which is an increase from the 4.2 million estimated by the WHO in 2016.70 Using novel hazard-response functions, it was recently estimated that up to 8.79 million global premature deaths could be attributed to air pollution every year.71 Non-communicable diseases, especially cardiovascular, are the major contributor to DALYs and global deaths, therefore, it is of great interest for public health that the interactions between air pollution and CVD is well understood.72
Air Pollution and Endothelial Function
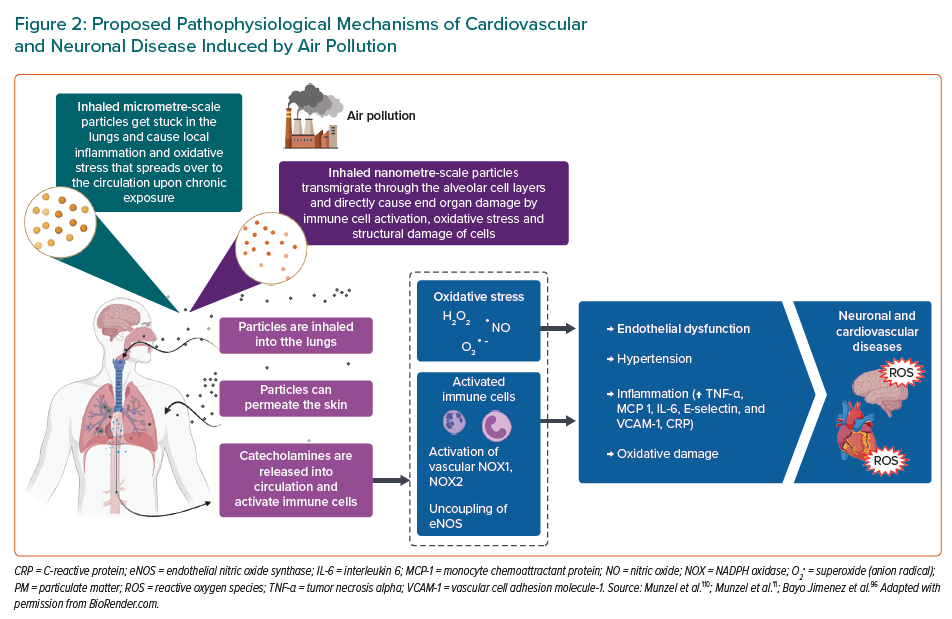
When inhaled, pollutants from the air need to reach the circulation to be able to interact with the endothelium. Gaseous components of air pollution can easily enter circulation, bypassing the air–blood barrier, where they can cause either direct damage by reacting with the constituents of the endothelium or indirectly by activating the immune system or causing oxidative stress.11,73 Nanometre-scale PM can also penetrate the air-blood barrier and directly migrate into the circulation.74,75 Larger, micrometre-scale PM usually don’t enter circulation, but exert their effects remotely, mostly by causing local inflammation that can later become systemic.76 Oxidative stress and inflammation are the two main risks that lead to onset and progression of endothelial dysfunction.
Oxidative stress can arise either from the free radical and ROS influx from air pollution itself or from the activation of ROS-generating enzymes in the body.77 One of the most prominent sources of ROS in the vascular tissue is the enzyme NADPH oxidase (NOX), which can either be activated in the vascular cells or come from circulating immune cells.78 The presence and activation of NOX from immune cells points to a close interplay between inflammation and oxidative stress, which often work together to progress endothelial dysfunction.79 A study done on Sprague-Dawley rats showed that exposure to concentrated ambient PM caused an impairment in the endothelium-dependent relaxation of isolated aortic rings, as well as activated the NOX in the vasculature.80 A similar study done on mice showed that impairment of the endothelium-dependent relaxation of isolated aortic rings was, in fact, mediated through oxidative stress caused by the upregulation of the monocyte-derived NOX.81 Exposure of animals to gaseous air pollutants was also shown to cause endothelial dysfunction. A study done on mice showed that exposure to ozone caused endothelial dysfunction that was accompanied by an increase in blood pressure and damage to mtDNA in aortic tissue.82 Other studies where animals were exposed to ozone also showed that presence of antioxidant enzymes like SOD and catalase can reverse the endothelial dysfunction.83 Exposure of Wistar rats to a combination of ozone and diesel PM caused upregulation of many different damage markers in aortic tissue, such as markers of oxidative stress (HO-1), of thrombosis (tissue factor and von von Willebrand factor), vasoconstriction (endothelin-1 and eNOS) and proteolysis (matrix metalloproteases-2 and -3).84 Chronic PM exposure of atherosclerosis-prone ApoE(-/-) mice was associated with increased accumulation of oxidatively modified cholesterol (7-ketocholesterol) and oxidised lipids in general, in the aortic plaques.85 The state of oxidative stress leads to direct effects on the vascular signalling through scavenging of NO, mostly by the superoxide radical.86 In rats exposed to ambient PM, it was shown that the impairment of the endothelium-dependent relaxation was associated with lower amounts of cyclic guanosine monophosphate (cGMP) and that NO donors did not improve the vascular function.87 Another study in rats, using diesel exhaust PM, showed that free radicals from the PM can directly scavenge NO, leading to impaired vascular signalling.88 Oxidative stress is not only detrimental to the central vascular system as pulmonary damage can influence pulmonary arteriole damage as well. Different chemical components of PM (e.g. heavy metals, reactive aldehydes, catalytic surfaces that directly produce ROS) can interact directly with the lung lining fluid and deplete antioxidants like glutathione and tocopherol, priming the lungs for oxidative damage.89 More recently, it was shown that also circadian dysregulation plays an important role for cardiovascular disorders caused by air pollution.66
Inflammation in the vasculature is not only a source of oxidative stress that impairs vascular signalling but is also responsible for onset and progression of diseases like atherosclerosis. Authors of a study on ApoE-/- mice observed that markers of inflammation TNF-α, MCP 1, IL-6, E-selectin and VCAM-1, were significantly elevated in the mesenteric arteries after exposure to concentrated ambient PM.90 Cardiac IL-6, TNF-α were also associated with 10 exposure of rats, pointing to direct inflammation of the cardiac tissue, and circulating levels of C reactive protein (CRP) pointing to systemic inflammation.91,92 It was also suggested that the inflammation could be mediated through the activation of the NLR family pyrin domain containing 3 inflammasome.93 There are also studies that identified inflammation as the main culprit for PM-induced endothelial dysfunction. In a study done on mice, dexamethasone, an anti-inflammatory substance, reversed the impaired acetylcholine-dependent relaxation of isolated aortic rings, but antioxidants did not.97 Gaseous components of air pollution are also responsible for inflammatory response, as shown in the meta-analysis that associated O3, NO2 and SO2 with increased CRP, but only associated NO2 exposure with elevated TNF-α.94 Most studies associate pulmonary exposure with ozone inhalation, but a study in rats has also demonstrated that systemic inflammation, envisaged by increased circulating IL-6, might accompany the observed endothelial dysfunction (explained in detail in Figure 2).95 In 2022, Wang et al. employed mice to assess vascular homeostasis following a standard urban particulate matter exposure, and they demonstrated that PM aspiration caused thickening of airways, leukocytes infiltration and adhesion to alveolus. In addition, the results showed an induction to vascular cell apoptosis and subcellular dysfunction, to which local and systemic redox biology and inflammation are probably correlated.97 Similar results were showed by Rojas et al., who exposed mice to a mixture of phenanthrene, fluoranthene and pyrene, which are compounds of PM. These exposed mice showed an increase in serum inflammatory cytokines and in expression of markers of endothelial dysfunction, both relevant characteristics associated with the onset of disease atherosclerosis and cardiovascular disease.98 For a selected overview of the mentioned experimental air pollution protocols in the context of endothelial dysfunction, see Supplementary Material Table 1.
Heavy Metal Pollution and Endothelial Dysfunction
Cadmium (Cd), lead (Pb) and mercury (Hg) are toxic elements commonly found in the environment, contributing to hypertension, as well as these toxic elements cause severe inflammation and oxidative stress.12,99–101 Cd is an especially toxic and persistent heavy metal and a known carcinogen.102,103 The major sources of Cd exposures are food, cigarette smoking and as a byproduct of industrial processes.104 It has been shown that Cd exposure is associated with hypertension, atherosclerosis, diabetes and nephropathy, all of which could be attributable to dysfunctional endothelial and smooth muscle cells.105 In 2015, it was demonstrated that subjects with chronic Cd exposure showed endothelial dysfunction since NO levels were lower in Cd-exposed subjects than non-exposed subjects. Additionally, plasma L-arginine decreased while asymmetric dimethylarginine (ADMA), an endogenous eNOS inhibitor, increased in Cd-exposed subjects.106 More recently, Liang et al. provided novel insight into understanding the alterations of lipid metabolism induced by Cd exposure in endothelial cells. They used human microvascular endothelial cells (HMEC-1) that were exposed to 40-μM Cd for 6, 12 or 24 hours or 10-, 20- or 40-μM Cd for 24 hours, respectively. The Cd exposure accelerated the decomposition of triglyceride and resulted in the accumulation of free fatty acids. These changes stimulated cytotoxicity, induced ROS generation, altered the mitochondrial membrane potential and decreased the ATP content, which eventually led to endothelial dysfunction and cell death.107 In vitro exposure of endothelial cells to Cd (15 μM) resulted in increased permeability, inhibition of cell proliferation and induction of cell death.108 Moreover, in animal studies, Cd induced hypertension, which involved oxidative stress and suppression of eNOS expression in blood vessels leading to impaired Ach-induced vasorelaxation.109 In 2013, it was demonstrated that acute Cd exposure may induce vascular injury through endothelial oxidative stress and increased vasoconstrictor activity by reducing NO bioavailability owing to the increased production of ROS by NADPH oxidase (explained in detail in Figure 3).110 However, further studies are needed to understand much better the pathway by which Cd affects endothelial cell function.111,112 Another key pathomechanism of Cd involves impairment of the circadian clock, also termed “cadmium chronotoxicity”.113
Environmental Pb exposure is an established risk factor for hypertension and a possible risk factor for cardiovascular disease mortality.99 Pb is a highly toxic metal in aquatic environments. Fish are at the top of the food chain in most aquatic environments and are the most susceptible to the toxic effects of Pb exposure.101 Not only is Pb ubiquitous in aquatic environments, but also high levels of Pb exposure can be caused by anthropogenic activities, including the manufacture of batteries, paint and cement, as well as mining and smelting.114 Exposure to Pb, a heavy metal highly toxic to humans when ingested or inhaled, contributed to 900,000 premature deaths, while toxic occupational hazards followed with 870,000 deaths.115 In a recent cohort including 14,289 adults aged 20 years or older who were enrolled in the Third National Health and Nutrition Examination Survey between 1988 and 1994, in 20% of the participants a concentration of Pb in blood of at least 5 μg/dl (≥0.24 μmol/l) was identified, while the geometric mean concentration of Pb in blood was 0.82 µg/dl in 2016.116 An increase in the concentration of Pb in blood from 1.0 to 6.7 μg/dl ([0.048–0.324 μmol/l]), which represents the tenth to 90th percentiles, was associated with all-cause mortality (HR 1.37, 95% CI [1.17–1.60]), cardiovascular disease mortality (HR 1.70, 95% CI [1.30–2.22]) and ischaemic heart disease mortality (HR 2.08, 95% CI [1.52–2.85]). The population attributable fraction of the concentration of Pb in blood for all-cause mortality was 18.0% (95% CI [10.9–26.1]), which is equivalent to 412,000 deaths annually. The results demonstrated that low-level environmental Pb exposure is an important risk factor for CVD mortality.117 Tubsakul et al. demonstrated that rats exposed to low level of lead acetate (100 mg/l) in the drinking water for 16 weeks induced had increases in blood pressure and peripheral vascular resistance, and decreases in blood pressure in response to intravenous infusion of acetylcholine. Additionally, rats exposed to Pb showed a downregulation of the eNOS and an upregulation of the NADPH oxidase expression.118 Interestingly, in 2016 an animal study showed an increase in Pb blood levels in rats exposed to lead acetate for 7 days (4 μg/100 g on the 1st day and 0.05 μg/100 g/day i.m. subsequently), as well as an increase in right ventricular pressures. They concluded that short-term and low-level exposure to Pb changes pulmonary hemodynamics and increases oxidative stress since vasodilatation to exogenous NO was reduced by Pb.119 More importantly, these studies have elucidated the cellular and molecular mechanisms of Pb action on cardiovascular/pulmonary systems, but a comprehensive strategy to prevent deaths from CVD should include efforts to reduce lead exposure.
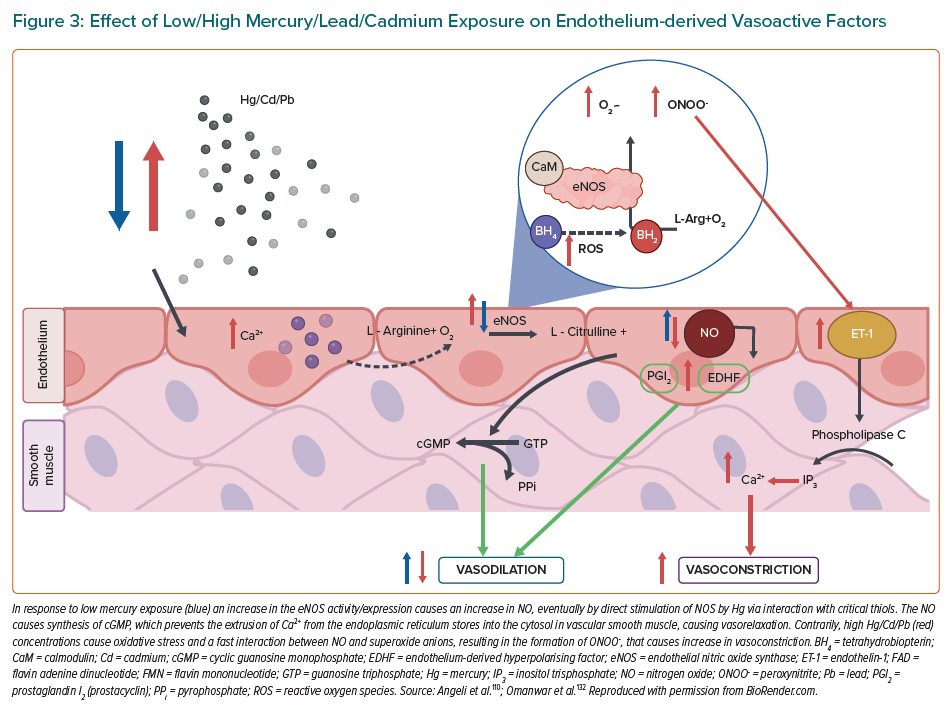
Global mercury emission inventories include anthropogenic emissions, contributing via current use or presence of mercury in a variety of products and processes, such as mercury-based artisanal and small-scale gold mining, as well as natural source emissions.120,121 Altogether, the release of this element into the environment leads to the pollution of air, water and soil. In addition, the inorganic form of mercury (Hg2+ ion) has deleterious effects in the kidney, whereas the methylated form of mercury (MeHg), which bioaccumulates in rice since its consumption is the primary pathway for methylmercury exposure at inland mercury (Hg) mining areas of China, targets the central nervous system (CNS) and has been classified as carcinogen since 1993.102,122,123 The most common route of exposure to mercury is through contaminated fish and other seafood.124 An association between the environmental and occupational exposure to mercury and the risk of CVDs was established after follow-up studies of severe cases of poisoning, and studies also showed cardiovascular abnormalities in Iraq and in Minamata, Japan.125,126 As it was introduced, endothelial dysfunction is characterised by a shift in the actions of the endothelium toward reduced vasodilation, a proinflammatory state and prothrombic properties. For this reason, there are some studies that associate chronic exposure to mercury and its derivatives with an increase of vascular resistance and an induction of hypertension.127–129 During the last years, it has been difficult to explain the definitive pathogenesis owing to the fact that the individuals exposed to mercury usually display multiple systemic disorders, including circadian clock dysregulation.113
For this reason, the proposed mechanisms are described by animal models. Golpon et al. demonstrated that mercury chloride (HgCl2) induces an endothelial-dependent vasorelaxation, which was totally blocked by the nitric oxide inhibitor L-NAME using norepinephrine preconstricted rat aorta and pulmonary artery rings with intact endothelium. Therefore, they concluded that HgCl2 alters structure and function of vascular endothelial cells.130 In 2014, similar results were reported that identified mercury exposure as causing a biphasic response: vasorelaxation at lower concentrations and vasoconstriction at higher concentrations. They determined that at low concentrations, mercury caused an endothelium-dependent vasorelaxation and at higher concentrations, mercury changes the composition and functionality of the vascular endothelium.131 These studies indicate that mercury produces a dual/biphasic response. In response to low mercury exposure, an increase in the eNOS activity/expression causes an increase in NO with no effect on prostacyclin (PGl2) and endothelium-derived hyperpolarising factor (EDHF). Both high and low mercury exposure also causes oxidative stress, and a fast interaction between NO and superoxide anions results in the formation of peroxynitrite (ONOO-), which causes vasoconstriction. However, in response to high mercury exposure, a decrease in the eNOS activity/expression causes a decrease in NO with persistence or upregulation of EDHF. Conversely, the endothelium is able to secrete the potent vasoconstrictor ET-1, which through its proinflammatory and mitogenic effects, augments the pathogenesis of CVD (Figure 3).133
Epidemiological/Observational Evidence on the Association of Environmental Risk Factors and Surrogate Markers of Endothelial Dysfunction
Air Pollution
There is mounting evidence from epidemiological/observational studies that exposure to various air pollutants, in particular to fine particulate matter (PM2.5), is associated with impaired endothelial function (Supplementary Material Table 2).11 In the Multi-Ethnic Study of Atherosclerosis (MESA)-Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution), long-term PM2.5 exposure was independently associated with a 0.3% decrease in flow-mediated dilation (FMD per interquartile increase in 3,040 subjects).134 Herein, no meaningful association was observed when investigating the short-term variation in PM2.5. A further study from the MESA cohort also indicated a positive association between PM2.5 and E-selectin.135 Likewise, analyses in 5,112 subjects from the framing cohort revealed that long-term PM2.5 exposure was associated with decreased FMD and hyperemic flow velocity.136 Weaker or no evidence for an association between endothelial dysfunction and PM2.5 was found in the Framingham cohort when investigating the hyperemic response measured by peripheral arterial tonometry.137 This result was also confirmed in a study by Ljungman et al.138 A meta-analysis of six studies by Sun et al. demonstrated that increases in vascular endothelial growth factor (VEGF) were associated with PM2.5 mass concentration.139 A more recent study (n=100) by Riggs et al. showed that a 12.4% decrease in reactive hyperemia index (RHI) was related to increased PM2.5.140 Moreover, PM2.5 was positively associated with VGEF and intracellular adhesion molecule-1 (ICAM-1), whereas a negative association was determined with vascular cell adhesion molecule-1 (VCAM-1). In a study investigating the interplay between endothelial function, oxidative stress and inflammation in young adults, episodic increases in PM2.5 exposure was related to higher levels of endothelial microparticles (annexin V+/CD41-/CD31+) along with increases in markers of endothelial adhesion, i.e. adhesion soluble intercellular adhesion molecule 1 (sICAM-1) and soluble vascular cellular adhesion molecule 1 (sVCAM-1).141 Data from 704 elderly men participating in the Veterans Administration Normative Aging Study suggested an association between short- and intermediate-term air pollution exposure (ozone (O3), particle number, black carbon, nitrogen dioxide (NO2), carbon monoxide (CO), sulfates and/or PM2.5) and ICAM-1 and VCAM-1.142 A study that further investigated the influence of different components of PM2.5 revealed that independent of PM2.5 mass vanadium was related to ICAM-1 and VCAM-1.143 In line, in a panel of 40 healthy adults from China, total PM2.5 from dust/soil and several crustal and transition metals were shown to be associated with increased endothelin 1 (ET-1) and ICAM-1 and decreased VCAM-1.144 Furthermore, CO was associated with higher ICAM-1, whereas nitric oxide (NO) was shown to be associated with decreased ICAM-1. Interestingly, endothelial dysfunction as displayed by increased levels of ET-1, E-selectin and ICAM-1 was shown to partly mediate the effects of personal exposure to PM2.5 components on blood pressure during pregnancy.145 In a recent study using data from the Heredity and Phenotype Intervention (HAPI) Heart Study, exposure to PM2.5 and PM10 was inversely associated with FMD.146
Noise Exposure
Recent comprehensive evidence on the association between noise exposure and endothelial dysfunction comes mainly from a series of field studies that was conducted by our noise research group. Here we could demonstrate that exposure to simulated night-time traffic noise is consistently associated with decreased FMD in healthy adults and patients at increased cardiovascular risk. In a first study, healthy participants were subjected to three different aircraft noise scenarios (0 overflights, 30 and 60 overflights) in a randomised manner.147 Night-time aircraft noise exposure led to decreased FMD (control group: 10.4%; noise 30: 9.7%; noise 60: 9.5%). In addition, pulse transit time was reduced after exposure to noise (control group: 271.8 ms; noise 30: 270.9 ms; noise 60: 264.9 ms), a parameter associated with increased blood pressure, vascular tone and stiffness. The noise-related deterioration of the FMD was particularly pronounced when the subjects were first exposed to the noise scenario 30 and then 60 simulated overflights, so that at the vessel level a noise-related sensitisation rather than a habituation could be observed (priming effect). Interestingly, the worsening of FMD in a small subgroup of subjects (n=5) exposed to noise scenario 60 could be improved by a single dose of vitamin C. This suggests that an increased formation of vascular reactive oxygen species may be at least partly involved in the pathogenesis of endothelial dysfunction. In a follow-up study, patients with present coronary artery disease or at increased risk were randomised to two different noise scenarios (60 overflights with a mean sound pressure level of 46.9 dB and a control scenario with usual ambient noise with a mean sound pressure level of 39.2 dB).148 Interestingly, the negative effects on endothelial function were more pronounced in patients with coronary artery disease (control group: FMD 9.6%; noise 60: FMD 7.9%) when compared to healthy subjects in the previous study. The comparison of 60 versus 120 overflights in a following study resulted in a FMD of 7.27 and 7.21%, respectively, compared with a FMD of 10.02% in the control night.149 The negative effects of simulated railway noise on endothelial function have also been demonstrated.150 Herein, simulated railway noise with a peak noise level of 65 dB and an average sound pressure level of up to 54 dB(A) (30–60 trains per night) led to a substantial deterioration of endothelial function in healthy subjects. As demonstrated previously, endothelial function could be significantly improved by the acute administration of vitamin C (2 g p.o.).
Conclusion
In the present review, we provide deep mechanistic insight into the molecular basis of noise, air, or heavy metal pollution-induced endothelial dysfunction. In 2019, the fourth largest risk factor for attributable deaths among males (ambient particulate matter and ambient ozone pollution accounting for 3.75 million deaths or 12.2 % of all male deaths in 2019) and females (2.92 million deaths or 11.3 % of all female deaths) was air pollution.69 There is also a wide range of other risk factors not yet included, such as nitrogen dioxide, heavy metals, environmental noise, sleep, mental (psychological) stress and UV radiation, among others. Furthermore, the WHO even estimates that up to 12.6 million deaths worldwide in 2012 were due to living in unhealthy environments, which means 24% of global deaths (and 28% of deaths among children under five) are due to modifiable environmental factors.151 Accordingly, there are numerous studies in the literature (summarised in Supplementary Material Tables 1 and 2) that have shown that the pollutants mentioned here cause oxidative stress and inflammatory effects on the cardiovascular system, impairing the health of the endothelium. It is known that a healthy endothelium is an important regulator of blood pressure both in terms of vascular tone modulation and as an interactive junction for the immune and cardiovascular systems. Undoubtedly, active research conducted on improved preclinical models associated with various pathways of protection against environmental risk factors, which could prevent the failure of the vascular function that contributes to the development of cardiovascular and neuronal diseases, as well as data obtained from the ongoing clinical research will provide answers to the currently open questions.