










Although significant improvements in the treatment of atherosclerotic cardiovascular disease (ASCVD), including early mechanical intervention and polypharmacotherapy with aggressive lipid modification, have been achieved in recent decades, there is still considerable residual cardiovascular (CV) risk.1 The latest trials have underlined the role of inflammation in CV disease (CVD), showing that lowering the inflammatory burden results in a reduction of future CV events.2–4 The aims of this review are to discuss the role of inflammation in CVD and to provide perspectives on future questions that need to be addressed with regard to treatment allocation.
Atherosclerosis as an Interplay Between Lipoproteins and Inflammation: Biology and Mechanisms
Canonically, atherosclerosis has been considered as a lipoprotein-driven disease, which is amplified and modified by the host immune cellular response to retained lipoproteins.5–7 The primary steps in early lesion formation are the accumulation and subsequent chemical modification (e.g. by lipolysis, proteolysis, glycation, aggregation or, most importantly, oxidation) of apolipoprotein B (apoB) containing lipoproteins, principally LDL in the subendothelial matrix with subsequent foam cell formation.8 Oxidised lipoproteins possess a variety of biological actions and consequences, including injury to endothelial cells, upregulation of the expression of adhesion molecules, recruitment and retention of leukocytes that recognise them as possible triggers of local inflammatory processes within the atherosclerotic plaque.
Interestingly, the potential of apoB lipoproteins to trigger inflammatory processes in the arterial wall may differ profoundly across apoB particles.7–10 Given this, certain metabolic conditions, such as an elevated hepatic triglyceride pool, or a genetic predisposition, such as LPA variants, significantly modulate the formation and/or metabolic disposition of atherogenic apoB lipoproteins, ultimately resulting in a more pro-inflammatory apoB phenotype.11,12 In addition, oxidised LDLs can both mimic damage-associated molecular pattern molecules (DAMPs), a well-known substrate for pattern recognition receptors on macrophages, and/or participate in the generation of intracellular crystalline cholesterol, thereby initiating an innate immune response through coactivation of a NLRP3 (NOD [nucleotide oligomerisation domain], LRR [leucine-rich repeat]-containing and PYD [pyrin domain]-containing protein 3) inflammasome within macrophages.13–16
In line with these mechanistic findings, observational evidence has demonstrated that elevated levels of circulating inflammatory biomarkers such as high-sensitivity C-reactive protein (hsCRP) or interleukin-6 (IL-6) independently predict the risk of ASCVD.17–20 Chronic low-grade inflammation accompanies all stages of atherosclerotic disease from its onset to the overt disease with manifest ischaemic syndromes, thereby potentially offering a new and important therapeutic option.
Inflammation as a Target for Intervention: Lipid-modifying and Anti-inflammatory Agents
The initial evidence that systemic inflammation can be modified pharmacologically stems from the lipid-lowering trials of the early 2000s, which demonstrated that statin therapy significantly reduced circulating levels of hsCRP, thereby confirming at least partially the interplay between inflammatory pathways and lipid metabolism.21–23 Since then, several landmark statin trials have clearly demonstrated that lowering hsCRP concentrations translated into a significant reduction of future CV events.24–26 Importantly, the reduction in hsCRP was found to be of similar magnitude to that seen with LDL-cholesterol (LDL-C) reduction. Additionally, these trials highlighted the fact that patients who demonstrated statin-associated reductions both in LDL-C to below 70 mg/dl and in hsCRP levels below 2 mg/l had a larger CV benefit than those who achieved substantial LDL-C lowering alone.24–26 Among other factors, this has led to the ‘residual inflammatory risk’ concept, defined as persistently elevated hsCRP despite sufficient atherogenic lipid lowering, a phenomenon seen among 30–40% of all statin trial participants.1
Interestingly, both a recent meta-analysis of 2,546 patients who were treated with a novel non-statin lipid-lowering drug class, namely PCSK9 (pro-protein convertase subtilisin-kexin type 9) inhibitors, as well as the two large PCSK9 inhibitor outcome trials FOURIER and ODYSSEY OUTCOMES demonstrated PCSK9 inhibitors had no significant effect on hsCRP despite profound LDL-C reduction (up to 50–60%).27–29 Nonetheless, post-hoc data from FOURIER and SPIRE in patients at high risk on statin treatment consistently documented that inflammation still plays an important prognostic role, even in subjects with very low LDL-C concentrations (<20 mg/dl), in whom hsCRP was able to independently modify CV risk.30,31 This supports the notion that additional anti-inflammatory treatment in these patients might provide benefit beyond aggressive lipid lowering.
To date, the pharmacological inhibition of various pathways involved in inflammation has been investigated in several clinical trials among patients with manifest ASCVD, who were already on statin therapy (>90% of all participants).32 Proof of principle that pharmacological lowering of persistent low-grade inflammation in the absence of lipid modification resulted in a reduction of incident ASCVD came from the CANTOS trial.2 This involved 10,061 stable patients with a history of MI and elevated baseline concentrations of hsCRP (≥2 mg/l) despite maximally tolerated statin treatment. Canakinumab, a fully human monoclonal antibody directed against IL-1β, not only significantly reduced concentrations of IL-6 and hsCRP by up to 40–60%, but was also associated with a significant reduction in the primary outcome (major adverse CV events [MACE] including non-fatal MI or stroke and CV death) overall by 15% and by 25% in those with on-treatment hsCRP level <2 mg/l.2,33 The CV protection achieved by canakinumab was identical in magnitude to that observed in major trials of PCSK9 inhibition (relative risk reduction of 15–20% over 2–3 years’ follow-up). In contrast, the results from the large CIRT study were rather disappointing.34 Here, 4,786 stable high-risk patients, either post-MI or presenting with multivessel disease and diabetes or metabolic syndrome on standard secondary prevention care, were randomly allocated to treatment with low-dose methotrexate versus placebo. Methotrexate therapy (15–20 mg weekly) neither decreased MACE over 5 years (HR 1.01; 95% CI [0.82–1.25]; p=0.91 versus placebo) nor lowered the concentration of inflammatory markers, particularly hsCRP.34
A third compound with potent anti-inflammatory properties tested in two large clinical trials was colchicine.3,4 In the COLCOT study, Tardif et al. demonstrated that colchicine at a low dose of 0.5 mg daily was able to reduce the risk of ischaemic ASCVD events (primary endpoint of death, resuscitated cardiac arrest, acute coronary syndrome, stroke and urgent hospitalisation for angina requiring revascularisation) by 23% compared with placebo in patients recruited in the first 30 days after an MI (n=4,755).3 When stratified according to time to randomisation, participants who received colchicine within 3 days of the index event sustained a 48% relative risk reduction in the primary endpoint.35
More recently, Nidorf et al. extended the COLCOT observations to patients with stable coronary artery disease (CAD) in the LoDoCo2 study (n=5,522).4 LoDoCo2, following the earlier open-label LoDoCo trial of low-dose colchicine involving only 532 patients with stable CAD, demonstrated – similar to COLCOT – a therapeutic benefit with colchicine 0.5 mg daily, with a 31% lower relative risk of the primary CVD endpoint compared to placebo after a median follow-up of 28.6 months.36
Unfortunately, neither COLCOT (measured in <3% of the study population) nor LoDoCo2 (not measured) provided sufficient insights into the inflammatory burden at baseline or on-treatment represented by hsCRP or IL-6 levels.3,4 Although colchicine seems to confer a clinical benefit in secondary prevention, clarification is still needed on optimal dosing and/or what therapeutic regimens should be chosen for optimal prevention of recurrent events, as discussed later.
NLRP3 Inflammasome: Linking Lipoproteins and Inflammation
One common feature of the above-discussed anti-inflammatory drugs for CV event reduction relates to their interaction with the canonical pathway of the NLRP3 inflammasome to IL-1/ IL-6/ CRP.15,16 Although a central action of colchicine and canakinumab is related to the inactivation of the NLRP3 inflammasome sequelae, these compounds target different components of the above mentioned pathway. Canakinumab has a specific mechanism of action, selectively targeting IL-1β and leaving IL-1α untouched.37 Colchicine, in contrast, has much broader effects on inflammation, predominantly as a consequence of the inhibition of tubulin polymerisation.38 However, its most important properties in the context of ASCVD are related to the suppression of caspase 1 activity, which prevents IL-1β cleavage from its precursor.39 In contrast, methotrexate has been shown to have no specific effects on the IL-1β/IL-6 pathway and to act via inhibition of aminoimidazole-4-carboximaide ribonucleotide with subsequent elevations in adenosine levels, and exhibited no reduction in CV endpoints.34,40 The results of the above trials suggests that inflammation related to atherogenesis may be pathway specific rather than a generalised inflammatory process. So far, it appears the NLRP3 inflammasome is linked to ASCVD.
Most importantly, these data further highlight activation of the NLRP3 inflammasome as a mechanistic link between vascular inflammation and the cholesterol pathway. Indeed, several components of lipid metabolism, such as oxidised LDL, cholesterol crystals, apoC3 and/or palmitic acid (C16:0) are pivotal regulators of the NLRP3-inflammasome either via dimerisation and activation of pattern recognition receptors such as toll-like receptors (TLR) 2 and TLR 2/4 or (especially in the case of cholesterol crystals) by lysosomal damage, enhanced potassium ion efflux, mitochondrial dysfunction and reactive oxygen species release (Figure 1).11,41–48
Once activated, the NLRP3 complex activates caspase-1, which, in turn, leads to IL-1 family cytokine production and the release of the highly proinflammatory cytokine IL-1β, the key mediator of atherosclerosis.16,49,50 IL-1β acts as a chemotactic agent for other inflammatory cells, resulting in a chronic, maladaptive inflammatory response dominated by macrophages and T-cells.7,8,15,49,50 Moreover, it induces the IL-6/IL-18 pathway and stimulates protein synthesis in the liver, which results in a characteristic shift towards an acute phase pattern characterised by elevated levels of not only CRP, but also serum amyloid A (SAA), fibrinogen, plasminogen-activator inhibitor-1 (PAI-1) and others.49–51 This vicious cycle sustains an inflammatory environment and mediates the ongoing cellular recruitment with the generation of foam cells and fatty streaks, eventually leading to the development of complex plaques.52 The seminal role of the NLRP3 inflammasome and the IL-1β pathway in atherosclerosis have recently been reviewed in detail.17,53,54
Lipoproteins, Inflammation and Thrombosis: a Dangerous Triad?
Viewed from another perspective, inflammation per se might be a key regulator of hepatic lipoprotein metabolism, supporting the concept of a bidirectional relationship.55 Lipoproteins such as apoC3-enriched very LDL (VLDL) particles and oxidised phospholipid (OxPL)-enriched lipoprotein(a) (Lp(a)) particles have the potential to trigger distinct inflammatory/immune responses.44,45,56 On the other hand, acute or chronic systemic inflammatory signals such as increased levels of tumour necrosis factor-α (TNF-α) cause rapid upregulation of lipid synthesis, secretion of VLDL particles into the plasma and reduced apoB degradation.57
This conserved lipoprotein response to infection and/or hyperinflammatory states may have served important evolutionary purposes in that the increased transport of lipids and the resulting hyperlipidaemia as part of the host’s protective response may have enhanced acute tissue repair after injury and fast energy delivery to tissues. In our current environment, it may rather be seen as a maladaptive response to chronic low-grade inflammatory conditions such as metabolic syndrome, visceral adiposity and diabetes, and may contribute to the vicious cycle of inflammation and dyslipidaemia.
One prominent example of a tight link between dyslipidaemia and vascular inflammation is illustrated by Lp(a). Lp(a) is an LDL-like particle with apo(a) bound covalently to apoB by a disulphide bridge.58 It has several features that render it more pathogenic and proatherogenic. Due to its homology with plasminogen, apo(a) has the prothrombotic properties of Lp(a).59 Furthermore, apo(a) has a distinct proatherogenic potential which is mainly attributable to its pro-inflammatory properties resulting in cytokine expression and release, as well as increased monocyte chemotaxis, which seems to be mediated in a large part by an enrichment in oxidised phospholipids (oxPLs).12,56 Indeed, Lp(a) is the major reservoir of oxPLs in human plasma.60,61
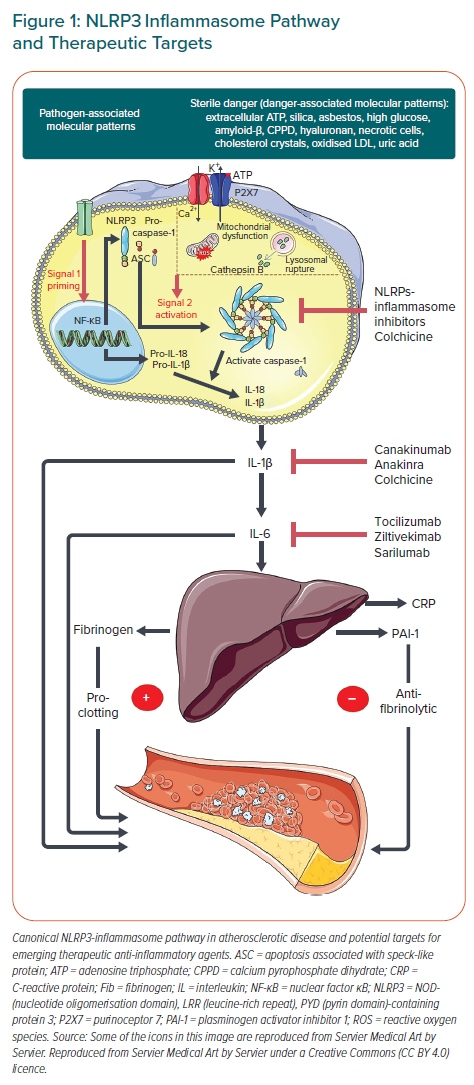
Moreover, recent ex vivo data has demonstrated that, on an equimolar basis, Lp(a) has much higher inflammatory potency than LDL-C.12 On the other hand, Lp(a) has been documented as an acute phase reactant, likely because the LPA gene contains an IL-6 response element, which suggests proinflammatory stimuli are involved in its regulation.62,63 The specificity of the IL-6 pathway in regulating Lp(a) production is further reflected by the fact that monoclonal antibodies directed against IL-6 (e.g. tocilizumab or sarilumab) reduced Lp(a) levels by 30–40%, whereas the antibody directed against TNF-α (adalimumab) did not affect Lp(a) levels substantially.64–66

Of interest, a prespecified secondary analysis of the ACCELERATE trial showed that elevated Lp(a) levels were associated with CV death, MI and stroke only in patients with hsCRP levels >2 mg/l but not in patients with hsCRP levels ≤2 mg/l.67 The interdependence of Lp(a) and systemic inflammation may have important clinical implications in terms of selecting patients who might benefit the most from Lp(a)-mediated ASCVD risk reduction, namely those with residual inflammatory risk. However, these results need to be confirmed by other studies. In general, subjects with elevated Lp(a) exhibit increased inflammatory activity in the arterial wall, as demonstrated by PET or CT.68 However, whether inflammation, at least partially, triggers Lp(a) production or Lp(a) induces an important immune response is still not entirely clear. The results from Puri et al. suggest an involvement of inflammatory stimuli in Lp(a) regulation, which is in line with the above-mentioned genetic regulation of Lp(a) in response to IL-6.67 On the other hand, treatment with an antisense oligonucleotide, a gene-based therapy aimed at silencing the translation of apo(a) that is associated with an approximately 80% Lp(a) reduction, was able to attenuate the pro-inflammatory state of circulating monocytes on the transcriptional as well as functional level in patients with elevated Lp(a), suggesting their reciprocal relation.69,70
Innate immunity is also tightly linked to a prothrombotic phenotype, a process now referred to as thromboinflammation.71 Several lines of evidence suggest an inflammatory response has a pivotal role in tissue factor expression, activation of platelets, hyperfibrinogenaemia and of PAI-I associated impaired fibrinolytic function (Figure 2).71,72 On the other hand, coagulation can also increase inflammation, thereby acting in a positive feedback loop.72 Interestingly, the recent introduction of platelet lipidomics reemphasised the role of circulatory lipids in inflammation-driven thrombosis.73 Several components of lipid metabolism, such as oxidised LDL and especially Lp(a), are thought to promote potent prothrombogenic activity.12,56,61 Moreover, rosuvastatin might have an additional pleiotropic effect beyond its anti-inflammatory properties by reducing platelet membrane cholesterol, as well as by diminishing levels of tissue factor, factor VII and factor X.74
In addition, a new player in lipidology, PCSK9, might also contribute to impaired platelet function and a procoagulatory state, as depicted by recent in vivo data.74 For instance, PCSK9 deletion in mice resulted in reduced formation of both arterial and venous thrombi, whereas overexpression of PCSK9 in septic mice promoted a hypercoagulable state, as reflected by elevated thrombin–antithrombin values in conjunction with reduced protein C.74
Circulating lipoproteins appear to affect both inflammation and thrombosis, although the exact mechanisms behind such complex interplay within this dangerous triad are still unclear.
Residual Cholesterol Versus Residual Inflammatory Risk: Rationale Applying a 2 × 2 Factorial Design for a Trial
As discussed, lipoprotein metabolism and low-grade inflammation are interrelated in their contribution to atherogenesis.54 Whether they act synergistically, potentiate their actions or have additional, independent effects still needs to be evaluated. However, there is clear evidence that using a combination of inflammatory and lipid parameters improves our ability to predict future ASCVD events.
In CIRT, which represented a contemporary, optimally treated population at very high risk, the combination of IL-6 (or hsCRP) and LDL-C resulted in an up to threefold better prediction of future MACE than did a single biomarker alone.75 Comparing the participants in the top quartile (Q) of IL-6 and the LDL-C distribution with those in the bottom quartile resulted in an adjusted HR of 6.4 (95% CI [2.9–14.1]). The risk estimate for increased hsCRP and LDL-C was similar at 4.9 (95% CI [2.6–9.4]). In contrast, HRs for a single biomarker (Q4 versus Q1, fully adjusted model) varied from 1.79 for hsCRP, 2.11 for IL-6 and 2.38 for LDL-C. These observations have significant clinical implications. First, they highlight the presence of both residual cholesterol risk and residual inflammatory risk despite aggressive, guideline-directed medical therapy, including statins and coronary revascularisation. Second, they indicate the equal importance of these two different types of residual risk. Finally, they support the need for a more comprehensive ‘dual pathway’ approach that goes beyond statins and simultaneously targets both dyslipidaemia and inflammation. The latter is of particular importance, since trials using a PCSK9 inhibitor or anti-inflammatory agents separately from each other resulted only in a 20–30% reduction in CV risk. This leaves considerable residual risk that needs to be addressed.
The important question is whether a combination of both aggressive lipid-lowering and anti-inflammatory therapy, which should be given only as ‘add-on’ therapy to optimal guideline-directed secondary prevention, not only might result in a more profound reduction of recurrent ASCVD events, but also may exceed the sum of the parts. Such an assumption can only be tested in the context of a cardiovascular outcomes trial employing a 2 × 2 factorial design. However, it is not clear yet how to optimally design such a clinical trial. Indeed, three central questions need to be answered first – what to use? who to use? and when to use? – reflecting drug selection, patient stratification and treatment modality (Figure 2).
Which Compounds to Select?
The first question to be answered by the research community is which drug/drug combination will provide an optimal choice? What is clear to date is that a combination of conventional lipid-lowering medication (maximally tolerated high-intensity statin and ezetimibe) with an anti-inflammatory drug would represent an ‘anti-inflammatory + placebo’ arm of the 2 × 2 trial (Figure 2; upper right panel of 2 × 2 table). However, the question remains as to which additional lipid-lowering therapy should be chosen for more aggressive LDL-C reduction (Figure 2; left panel of 2 × 2 table), particularly as the trial design would employ significantly lower LDL-C thresholds than those in current guideline recommendations.
Would compounds antagonising PCSK9 or bempedoic acid be a promising approach for this purpose? A clear limitation of PCSK9 inhibitors is their cost but their negligible effect on hsCRP concentration make them ideal candidates for an unbiased estimate of 2 × 2 effects on risk reduction. A combination of bempedoic acid with ezetimibe, which has been recently approved, might have LDL-C lowering effects only similar to moderate intensity statin therapy; moreover, CV outcomes data are still awaited.76 Another possible choice among forthcoming novel lipid-lowering agents might be inclisiran, a small interfering RNA molecule that reduces hepatic PCSK9 production by approximately 50%.77 In addition, inclisiran administration twice yearly might be a major advance in lipid-lowering strategies.78
There is unequivocal evidence that direct inhibition of the inflammatory pathway targeting the NLRP-3 inflammasome has the potential to become a cornerstone therapy for atherosclerotic disorders. However, in contrast to a plethora of compounds used for lipid management, only two anti-inflammatory drugs – canakinumab and colchicine – have been shown to improve cardiovascular outcomes. To date, there has been no head-to-head comparison between canakinumab and colchicine and whether such a trial would ever be conducted is questionable, especially taking into account the huge differences in treatment costs and route of administration (oral daily versus subcutaneous every 3 months). In general, colchicine, being an oral, well-tolerated, rapid-acting and, most importantly, highly cost-effective drug compared to canakinumab might be the only feasible anti-inflammatory compound to date to be used in a 2 × 2 factorial trial.79
Its further advantage may be related to its broader mechanism of action compared to canakinumab. For instance, the recently published LoDoCo2 proteomic substudy demonstrated that 30-day colchicine treatment reduced not only the level of NLRP3 inflammasome-related cytokines, such as IL-1/IL-6/IL-18, but also resulted in the reduction of 11 inflammasome-unrelated proteins, such as myeloblastin, carcinoembryonic antigen-related cell adhesion molecule 8, azurocidin and myeloperoxidase.80 In addition, upregulation of 23 biomarkers with potent anti-atherosclerotic effects, such as fibroblast growth factor and insulin-like growth factor-binding protein, has also been shown.80 However, whether these additional beneficial effects of colchicine might result in a more pronounced event reduction than that seen in canakinumab therapy alone remains unknown.
Recently, Ridker et al. showed that patients on canakinumab were still at substantial residual inflammatory risk.81 Despite significant IL-6 lowering (43.2% from baseline on a 300 mg dose of canakinumab), each tertile increase in IL-6 concentration, measured 3 months after canakinumab initiation, was associated with a 42% higher risk for MACE (95% CI [26–59%]; p<0.0001). Weaker results have been obtained for IL-18 (15% increase in risk; 95% CI [3–29%]; p=0.016), although canakinumab did not alter IL-18 levels at 3 months significantly.
Do we need novel anti-inflammatory agents that affect the upstream NLRP3-inflammasome, reducing both IL-1β and IL-18? Theoretically, yes, and there are several NLRP3-inflammasome inhibitors already under development.82 However, more safety data are needed, since such profound, systemic inhibition of inflammatory pathways always bears potential risks due to interaction with immune homeostasis.
In CANTOS, direct targeting of IL-1β by canakinumab was associated with a small but statistically significant risk for fatal infections (0.31 versus 0.18 events per 100 person-years; p=0.02).2 Whether targeting a downstream molecule of the NLRP3-inflammasome by directly inhibiting IL-6 is a better solution – especially taking into account that IL-6 is causally involved in atherogenesis, as shown by Mendelian randomisation analysis – is an issue of ongoing investigation.83
Currently, several IL-6 pathway inhibitors are under investigation, including a monoclonal antibody against IL-6 (e.g. tocilizumab) or its receptor (e.g. sarilumab).51,84 Furthermore, ziltivekimab, a human monoclonal antibody targeting IL-6 is being evaluated in the Phase IIb study, RESCUE (NCT03926117), which is testing the value of inflammation reduction in patients with chronic kidney disease. This is of particular importance as colchicine is contraindicated in patients with chronic kidney disease, which indicates a clear need for an alternative anti-inflammatory drug for the subgroup with renal dysfunction.
Which Patients Might Benefit Most?
Selection of patients is critical. Should LDL-C levels be reduced to less than 55 mg/dl prior to initiating anti-inflammatory therapy? Furthermore, should anti-inflammatory therapy be applied to all ASCVD patients or only to those with residual inflammatory risk? Certainly, from a pathophysiologic standpoint, this would make sense and has been shown to work in JUPITER and CANTOS.2,26
However, it is not clear as to which biomarkers would be the best to use to identify those at high risk. The CANTOS trial included only patients with a high residual inflammatory risk, reflected by a hsCRP-concentration above 2 mg/l despite statin treatment.2 In contrast, the COLCOT trial did not use hsCRP as an inclusion criterion although, in a relatively small subset of patients, the median concentration was found to be very similar to that in the CANTOS trial at about 4 mg/l.3 Whether this hsCRP concentration, measured in such a small subset of patients, is representative of the hsCRP concentration in a trial population is not known. In CIRT – a negative trial that did not require an elevated hsCRP for inclusion – the median hsCRP concentration was much lower than in CANTOS (1.5 mg/l at randomisation).34 Nonetheless, a ‘responder’ analysis from CANTOS provided evidence that those who achieved a hsCRP of less than 2 mg/l benefited most from anti-inflammatory treatment, with a 25% reduction in MACE (HR 0.75; 95% CI [0.66–0.85]; p<0.0001), whereas in ‘non-responders’ only marginal treatment effects were observed (HR 0.90; 95% CI [0.79–1.02]; p=0.11).33 Even more convincing data were obtained for IL-6 ‘responders’ who achieved on-treatment IL-6 levels below 1.65 ng/l and demonstrated a 32% reduction in MACE.33
When to Treat and How Long to Treat?
Other issues that need to be addressed are related to the regimen of anti-inflammatory treatment such as time of treatment initiation or duration (life-long, e.g. aspirin and statins, versus intermediate- or long-term, e.g. P2Y12 receptor inhibitors). Are time-dependent treatment effects to be expected? Will targeting inflammation translate into improved cardiovascular outcomes in the short term or will the impact of this approach reveal its full potential over the long run? Are we dealing with permanent suppression of inflammation or should we anticipate a recurrence in MACE after cessation of therapy? We still do not know how long anti-inflammatory treatment needs to be continued and further research in this direction is urgently needed.
More recently, a secondary analysis of the CANTOS trial revealed intriguing results.85 Regarding canakinumab therapy, Everett at al. assessed the total (initial and subsequent) number of CV events, since patients remained on their assigned treatment for the total duration of the trial, even if they experienced a primary endpoint.85 In contrast to the primary analysis, a significant reduction of total major CV events was seen among all therapeutic regimens, including patients who were randomised to the lowest dose of canakinumab (RR 0.80; 95% CI [0.69–0.93]; RR 0.79; 95%CI [0.68–0.92]; and RR 0.78 (95% CI: 0.67–0.91) for 50 mg, 150 mg and 300 mg treatment groups, respectively. Moreover, significant benefits of anti-inflammatory therapy were demonstrated even among subjects who did not achieve a hsCRP concentration below 2 mg/l or an IL-6 concentration below 1.65 ng/l.
Interesting results have been also provided by the COLCHICINE-PCI study, which tested a strategy of preprocedural administration of a much higher colchicine dose of 1.8 mg among 400 subjects with chronic or acute PCI.86 Although an acute rise in IL-6 and hsCRP could be prevented by short-term colchicine administration, no significant risk reduction in PCI-related myocardial injury/MI or MACE within 30 days was seen.
Theoretically, the data above might favour prolonged anti-inflammatory treatment, at least among those at high risk for recurrent CV events (e.g. the PEGASUS trial) but, in practice, we are still far away from having solid data on the required duration of anti-inflammatory treatment.87
Another question related to treatment modality is: at what stage of the disease (acute or chronic) would targeting inflammation be most beneficial? Canakinumab was investigated only among patients who had experienced a documented MI at least 30 days before randomisation, whereas colchicine demonstrated its efficacy in those with acute MI as well as in patients with stable CAD.2–4 On the other hand, a time-to-treatment initiation (TTI) analysis within COLCOT highlighted the importance of early initiation of colchicine therapy for favourable CV outcomes after MI.35 Patients in whom treatment with colchicine was initiated within the first 3 days of the index event demonstrated a profound 48% reduction of the composite primary endpoint (HR 0.52; 95% CI [0.32–0.84]; p=0.007) compared to patients whose treatment started within 4–7 days (HR 0.96; 95% CI [0.53–1.75]; p=0.896) or >8 days (HR 0.82; 95% CI [0.61–1.11]; p=0.200), respectively.
While the role of combined lipid-lowering and anti-inflammatory therapy might become a cornerstone in the prevention and treatment of ASCVD, it is also prudent to promote lifestyle modification that might have anti-inflammatory potential.88 A recent simulation study revealed that optimisation of modifiable lifestyle risk factors such as body weight, smoking, physical inactivity and diet would shift almost 40% of patients with CAD and a high inflammatory burden beneath the considered treatment threshold of hsCRP >2mg/l.89 The ketone body ß-hydroxybutyrate (BHB), an endogenous inhibitor of the NLRP-3 inflammasome, is a promising molecule in this regard. Its physiological and transient elevation can be achieved by vigorous physical activity, (intermittent) fasting and dietary carbohydrate restriction.90,91 BHB promotes its anti-inflammatory properties by inhibiting the assembly of the NLRP-3 inflammasome and further reduction of expression of its downstream mediators (i.e. the IL-1β/IL-6 axis).83 In line with this, low carbohydrate dietary patterns that result in nutritional ketosis have been shown to reduce broad, systemic inflammation and most biomarkers of ASCVD risk in the insulin-resistant phenotype.92 Overall, elevated levels of BHB in plasma seem to shift tissue cross-talk from a proinflammatory milieu conducive to high-risk atherosclerosis to an anti-atherogenic milieu so might be a promising non-pharmacological therapeutic avenue for addressing residual inflammatory risk.
Conclusion
To date, varying inflammatory and/or atherogenic potential across different apoB phenotypes is not fully addressed by strategies directed at apoB/LDL-C lowering alone and results in residual inflammatory risk despite optimal lipid-lowering therapy. This highlights the importance of moving beyond assessment and treatment of a single marker such as apoB/LDL-C to comprehensively address all drivers of atherosclerotic risk, including inflammation. Pharmacologically, a variety of compounds are under investigation, primarily those targeting the NLRP3-IL1-β/IL-6 pathway, which might be best tested in high-risk post-MI patients, optimally identified on the basis of an increased inflammatory burden. However, time of initiation and duration of treatment have still to be determined. As discussed in this review, a large trial based on a 2 × 2 factorial design applying aggressive LDL-C lowering and aggressive anti-inflammatory (anticytokine) treatment to fully evaluate the potential additional value of reducing inflammation in the presence of ultra-low LDL-C concentrations may further optimise outcomes. Moreover, it may be important to go beyond these targets and simultaneously address residual risk related to elevated triglycerides, elevated Lp(a) and thrombotic burden. This comprehensive approach to residual risk management has the potential to revolutionise the prevention of cardiovascular disease. Nonetheless, we are still at the beginning of this journey with plenty of questions that need to be adequately addressed.