







Elevated levels of low-density lipoprotein cholesterol (LDL-C) have long been established as one of the most important risk factors for developing coronary artery disease (CAD) and other forms of atherosclerotic cardiovascular disease (CVD).1,2 Targeting LDL-C reduction has been effective in lowering cardiovascular risk.3,4 The use of HMGCoA reductase inhibitors (statins) for both primary and secondary cardiovascular disease prevention has become first-line therapy and has resulted in reduction of cardiovascular (CV) events and overall mortality.3 More recent guidelines from the American College of Cardiology (ACC) and American Heart Association (AHA) have recommended intensification of statin therapy based on calculated atherosclerotic CVD risk and not based on LDL-C levels alone.5 However, despite reduction in LDL-C by 18–41 % (moderate doses) and 40–60 % (higher doses or more potent statins), there remains significant residual cardiovascular risk. In addition, there are many other individuals that have been unable to achieve sufficient LDL-C lowering or have been intolerant to the drug completely.6 Other cholesterol lowering agents (bile acid sequestrants, ezetimibe, niacin, fibrates) have been used in conjunction with statins to achieve historic LDL and/or non-HDL-C goals, however at this time their addition has not been proven to have added value in clinical cardiovascular outcomes.7–10 Due to continued inability to achieve adequate clinical benefit and ongoing incidence of statin intolerance, research for alternative therapies is of utmost priority. The discovery of proprotein convertase subtilisin/kexin Type 9 (PCSK9) has opened the possibility for effective and adjunctive therapy for those who are not optimised with statins while providing therapy in lowering cardiovascular risk in subjects who are intolerant and have little alternatives.
LDL Cholesterol Cellular Metabolism
Most LDL is cleared from the plasma by LDL receptors on hepatocytes. At neutral pH, the LDL-receptor (LDL-R) binds to apolipoprotein B-100 (apoB100) on LDL and LDL endocytosis ensues.11 In the acidic environment of the endosome, the LDL-R folds into a closed formation state, thereby releasing LDL.12 LDL-R is then recycled to the cell surface while LDL is transferred to a proteasome targeting it for further processing.13 The inability of LDLR-LDLP complex to dissociate routes the complex for degradation, thereby reducing LDL-R surface concentration. Regulation of the LDL-R is also driven by the intracellular concentration of cholesterol. In the setting of high intracellular cholesterol levels, LDL-R levels decrease and activity of HMG CoA synthase and reductase decrease by more than 90 %. Thus, in the setting of drugs that lower intracellular cholesterol, such as statins, LDL-R levels are increased and serum LDL-C levels are decreased.
B Cellular mechanism regulating cholesterol homeostasis is complex and involves balance between intracellular biosynthesis and exogenous uptake of cholesterol. Cholesterol uptake and intracellular biosynthesis is regulated by sterol regulatory element-binding protein (SREBP), a transcription factor located in the endoplasmic reticulum. When cholesterol levels on the endoplasmic reticulum membrane are low, a sterol sensor binds to SREBP and signals its movement from the endoplasmic reticulum to the Golgi apparatus where SREBP is further activated and transported to the nucleus.14 In the nucleus, SREBP acts as a transcription factor, increasing HMG coA reductase and LDLR gene expression.13
Genetics of PCSK9
PCSK9 was first discovered as a ninth member of the subtilase subfamily,15 Its function was initially largely unknown until its role in hypercholesterolaemia was realised in a group of French families with autosomal dominant familial hyperlipidaemia (FH). These individuals did not exhibit the usual genetic mutations in the LDLR (LDL receptor) or APOB (apolipoprotein B) genes.16 Instead, the genotype of these FH cohorts revealed multiple missense mutations in the PCSK9 gene resulting in defective intracellular PCSK9 processing (see Table 1). A gain of function mutation results from the defective pro-PCSK9 processing and causes either an over expression of PCSK9 or an increased affinity for the LDL receptor.17 These gain of function mutations have shown to significantly decrease LDL receptors by as much as 35 % and consequently increase serum LDL-C levels.18 Targeting PCSK9 as a therapeutic strategy was conceptualised after loss of function mutations were discovered in both African American and Caucasian populations (see Table 1).19,20 Subjects with low LDL-C in the Dallas Heart Study had PCSK9 sequencing that identified two non-sense mutations in 2 % of the African American cohort resulting in low levels of PCSK9 and a marked reduction of plasma LDL-C of 40 %.21 Similarly, missense mutations were also discovered in the Caucasian population that was associated with 30 % decrease in LDL-C.19 Subjects with loss of function mutations and its association with ischaemic heart disease have been evaluated in a meta-analysis. In multiple groups of subjects with loss of function mutations in the PCSK9 gene, there was a 13 % decrease of LDL-C that was associated with 30 % risk reduction in incidence of ischemic heart disease.20 The higher than expected CVD risk reduction is thought to be from life-long exposure to very low LDL-C levels.

Molecular Mechanism of PCSK9
PCSK9 is a serine protease that belongs to a subfamily of subtilisin proteases responsible for activation or inactivation of other proteins, such as hormones, growth factors and other enzymes. It was initially identified as a seceretory proprotein convertase neural apoptosisregulated convertase-1 (NARC-1) prior to the discovery of its role in cholesterol metabolism.15 PCSK9 proprotein is processed in the endoplasmic reticulum where it undergoes autocatalytic cleavage producing a cleaved, but bound, prodomain and catalytic subunit. The bound prodomain makes the catalytic domain enzymatically inactive, unlike other serine proteases, acting instead as a binding protein when secreted. In the extracellular space, PCSK9 binds to LDL receptor via its catalytic subunit while its C-terminal subunit acts as a chaparone for endocytosis and shuttling the PCSK9-LDLR-LDL complex to lysosomes for degradation.22 In the absence of PCSK9 and in the acidic environment of the clathrin coated endosome, LDLR-LDL complex dissociates and LDLR is recycled back to the cell surface (see Figure 1).23
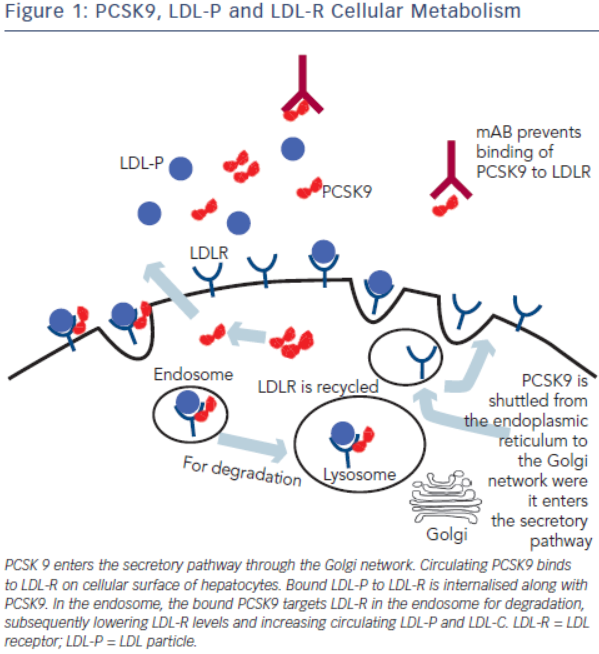
The PCSK9 gene is located on chromosome 1p32 and its expression is also regulated by intracellular cholesterol, via SREBP-2.24 Synthesis of PCSK9 occurs mostly in the liver, small intestine and kidney and is secreted as a soluble enzyme.25 After secretion, 30–40 % of PCSK9 binds to LDL, a process that may inhibit its uptake of LDL by LDLR.26 Low intracellular levels of cholesterol not only stimulate the synthesis of LDL-R but also PCSK9, serving as a counter-regulatory mechanism to maintain intracellular delivery of cholesterol. In the setting of statin, fibrate and ezetimibe use, PCSK9 expression is up-regulated due to low intracellular cholesterol levels.27 The degree of PCSK9 expression correlates with degree of LDL-C levels in subjects treated with statin, attenuating the cholesterol-lowering effects of statin.28 Thus, PCSK9 inhibition is additive to statin therapy and play a synergistic role in its lipid-lowering effects.
Inhibition of PCSK9
PCSK9 inhibition is currently achieved by either preventing PCSK9 synthesis or inhibiting the binding of PCSK9 to LDL-R. The most clinically advanced mechanism by which inhibition of PCSK9 has been achieved is through the development of monoclonal antibodies. Other mechanisms, such as mimetic peptides, antisense RNA inhibition and natural binding inhibitors are mostly in preclinical development (see Table 2).
Monoclonal Antibodies
In 2009, the first successful development of monoclonal antibody against PCSK9 was developed by Chan et al.29 The fully human monoclonal antibody (mAB) binds to both the catalytic and prodomain sites preventing PCSK9 binding to LDL-R.29 Monoclonal AB was shown to increase LDL receptor levels by 1.7-2.2-fold and had synergistic effects when administered concomitantly with statin.29 Phase I studies showed significant efficacy in reducing LDL-C levels with minimal adverse events.30,31 Almost simultaneously, multiple monoclonal antibodies have been developed with rapid completion of phase II trials and now ongoing phase III clinical trials.32–34
Alirocumab (REGN727/SAR236553)
Initial phase I clinical trials of alirocumab showed a dose-dependent LDL-C lowering effects. In one phase I clinical trial, escalating subcutaneous doses of 50, 100, and 150 mg doses were compared to placebo, lowering LDL-C by 39, 54 and 61 %, respectively (see Table 3).31 Phase II clinical trials have evaluated alirocumab’s use in heterozygous FH and in subjects with hyperlipidaemia on either stable doses of statin or escalating doses of statin (low and high dose atorvastatin). In the phase II clinical trial evaluating the use of alirocumab in the background of stable doses of atorvastatin (low or high dose) in subjects with LDL-C ≥ 100 mg, LDL-C decreased by as much as 72 % with 150 mg administered every two weeks.35,36 This study also showed significant decreases in apolipoprotein B (apoB), non-high density lipoprotein cholesterol (non-HDL-C) and lipoprotein (a) [Lp(a)].35 The mechanism by which Lp(a) is lowered is unknown, but maybe related to the decrease in the availability of LDL-apoB [known to decrease Lp(a)] and the increased clearance of apolipoprotein(a).37
In the phase II clinical trial evaluating the safety and efficacy of escalating dose of alirocumab in subjects with heterozygous FH, alirocumab was given either every two (Q2 week) or four (Q4 week) weeks in the background statin use with or without ezetimibe.38 Alirocumab was well-tolerated and effective in adequately treating subjects with FH (68 % reduction in LDL-C with 150 mg SC dose Q2 weeks compared with 11 % in the placebo group).38 The ODYSSEY program is currently underway and is a collection of 14 clinical phase III trials evaluating the efficacy and safety of alirocumab in various patient populations and clinical settings. All trials in the program are designed to treat to goal levels of LDL-C and evaluates efficacy in patient population that are not optimised with current standard therapies. This program involves over 2,000 international sites with over 23,500 subjects.39 The largest study will be evaluating clinical cardiovascular outcomes (ODYSSEYOUTCOMES), which is enrolling subjects with prior history of acute coronary syndrome already on optimised-guideline based therapy.40 In the recent European Society of Cardiology (ESC) scientific meeting, preliminary results from the ODYSSEY program were presented. Particularly intriguing was a post-hoc analysis of the ODYSSEY LONG TERM trial evaluating LDL-C lowering effects of alirocumab in patients with heterozygous FH or patients with high CV risk. Not surprisingly, alirocumab was shown to decrease LDL-C levels by 61 % compared to placebo. However, after only one year of the intervention post-hoc analysis showed a lower risk for CV events (HR 0.46, CI 0.26-0.82, p = <0.01) (ESC Barcelona Spain 2014).
Evolocumab (AMG 145)
Evolocumab, a full human monoclonal antibody, is administered subcutaneously either as every two weeks or every four weeks dosing regimen. Koren at al. showed that escalating doses of evolocumab as a monotherapy in both Q2 week or Q4 week dosing administered subcutaneously to subjects with history of hypercholesterolaemia and low cardiovascular risk (Framingham risk score < 10 %) decreased LDL-C from baseline by up to 48 %.41,42 Stein et al. in the phase II Goal achievement after utilising an anti- PCSK9 antibody in statin intolerant subjects (GAUSS) evaluated the efficacy and safety of evolocumab in patients with statin intolerance.43 Patients receiving escalating doses experienced a significantly reduced LDL-C compared to placebo. At 280 mg dose, LDL-C was reduced by 41 %.43 When escalated to a dose of 420 mg, LDL-C was reduced by 51 %.43 This study was expanded to a phase III trial in subjects with a history of hypercholesterolemia not at previously established National Cholesterol Education Program Adult Treatment Program III (NCEP-ATP III) risk-based goals and intolerant to statins.44 As in the phase II results, any regimen administered significantly reduced LDL-C compared to ezetimibe, with the highest reduction (61 %) seen with evolocumab 420 mg + ezetimibe 10 mg. When administered to subjects who were already taking statin, administration of evolocumab further decreased LDL-C by 63–75 % compared to placebo.45 The use of evolocumab was also studied in subjects with heterozygous FH already on maximized tolerated lipid therapy (stable dose of statin with or without ezetimibe).46,47 As in other studies, the largest additional decrease in LDL-C was seen with dose of 420 mg Q4 week (55 % additional LDL-C reduction).44 In subjects with known CAD or CAD risk equivalent and not at NCEP-ATP III goal of LDL-C ≤ 70 mg/dL, evolocumab was studied as an adjunct to stable doses of statin with or without ezetimibe.48 When administered in escalating doses either as a Q2 week or Q4 week regimen significant reductions in LDL-C occurred. The Q2 week regimen at 140 mg, achieved the highest LDL-C reduction at 12 weeks (64 %). Each dosing group also significantly achieved NCEPATP III treatment goal when compared to placebo (90 % of those in the Q2 week 140 mg group achieved LDL-C ≤ 70 mg/dL).48 An analysis from this study also showed therapy with evolocumab decreased Lp(a) by 23 % compared to placebo.49 As previously discussed, mechanisms by which this effect occurs is unknown.
The use of evolocumab was also studied in homozygous FH, a rare but severely elevated LDL-C, wherein very high CVD risk with possible death in childhood occurs. In the small study by Stein et al., the effect of evolocumab was studied in both LDL-R negative subjects and LDL-R defective patients. Evolocumab significantly reduced LDL-C by 26 % in only the LDL-R defective subjects. In a larger, randomised controlled trial that enrolled homozygous FH, evolocumab consistently decreased LDL-C by 23 %.50
There are currently phase III clinical trials that have shown safety and efficacy in lowering LDL-C with evolocumab. In a 12-week phase III clinical trial, evolocumab use in combination with moderate or high-intensity statin showed significant reduction in LDL-C (up to 75 % reduction when administered every two or four weeks).45 Another phase III study evaluating evolocumab use as a monotherapy also showed significant decrease in LDL-C (57 % more reduction compared to placebo and 40 % more reduction than ezetimibe).42 In a 12-week phase III study evaluating evolocumab use in subjects with intolerance to statin, those treated with evolocumab had significant reduction of LDL-C compared to ezetimibe (53–56 % vs 37–39 % p< 0.001).44 Patients that were previously enrolled in prior phase II studies (GAUSS, RUTHERFORD, LAPLACE-TIMI 57, and MENDEL) were evaluated in the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) trial. This study showed that subjects who continued to take evolocumab for the duration of the year on a monthly dosing regimen, maintained decreased LDL-C levels, whereas those that discontinued the study drug resumed their baseline levels.51 The Further cardiovascular outcomes research with PCSK9 inhibition in Subjects with Elevated CV Risk (FOURNIER) is currently enrolling to evaluate CV outcomes with evolocumab use in subjects already on guideline-based lipid therapy.
Bococizumab (RN316/PF04950615)
The monoclonal antibody bococizumab has undergone phase I clinical trial and shown that single and escalating intravenous and subcutaneous dose significantly lowered LDL-C by as much as 54 % with 150 mg Q2 week regimen without significant adverse effects.52 Currently, two phase III trials are underway studying bococizumab effects on cardiovascular outcomes in high risk subjects. In one of these outcomes trials, bococizumab use will be evaluated in subjects who are at high risk and have an LDL-C level of 70-100 mg/dL (SPIRE1). In another CV outcomes study, safety and efficacy of bobocizumab will be evaluated in high risk subjects who have higher than LDL 100 mg/ dL, despite high-dose statin or at the highest tolerated dose (SPIRE2) (see Table 4).
Other Targets for PCSK9 Inhibition
Other pharmacologic approaches to PCSK9 inhibition have been proposed; many are still in preclinical development. Direct inhibition of PCSK9 can be attained using small mimetic peptides called adnectins. Mimetic peptides of the PCSK9 binding domain for LDL have been shown to decrease LDL-R degradation.53,54 Mimetic peptides of the binding domain of LDL-R can also bind to PCSK9 and prevent its interaction with LDL-R.53,55 Another approach in PCSK9 inhibition is gene silencing techniques. Antisense oligonucleotides (ASO) are in preclinical development. ASO administration in mice has been shown to increase hepatic LDL-R receptor concentration.56 Antisense RNAs (siRNA) are also being developed to target PCSK9 mRNA, shown in non-human primates to reduce LDL-C by up to 50–70 percent.57 Natural inhibitors, such as annexin A2, a protein expressed in many tissues, inhibit PCSK9 and increase LDL-R.58,59 Berberine, a natural occurring plant alkaloid has been shown to also decrease PCSK9 mRNA expression and increase LDL-R in vitro and animal studies.60,61
Adverse Events
Serious adverse events from monoclonal antibodies targeting PCSK9 are rare. The most common adverse reactions are local injection site reactions. In the GAUSS phase II trial evaluating subjects that have intolerance to statin, myalgias were the most common adverse event but had low incidence overall.43 Alirocumab had similar adverse reactions between placebo and treatment groups in its phase II trials. In a dose escalating study of alirocumab, one of 152 subjects receiving a dose of alirocumab developed cutaneous leukocytoclastic vasculitis that was successfully treated with prednisone.35 Most common adverse reactions that caused discontinuation of medication in these trials were mild injection site reactions (erythema, pruritis, discoloration, haematoma, swelling). For evolocumab, the most common treatment related adverse reaction was not only injection site reaction (pain), but also headache and skin burning sensation.46
Conclusion
PCSK9 Inhibition for Use in Cardiovascular Disease Prevention
Although statins have revolutionised lipid therapy and secondary cardiovascular disease prevention, there remains a significant residual risk that can be further targeted. Certain populations may benefit from additive therapies: those with persistent elevation of LDL-C or residual risk despite optimal guideline-based therapy. Subjects with familial hypercholesterolaemia, for example, have been difficult to treat; despite maximum doses of lipid therapies, many subjects may not achieve sufficient reduction of LDL-C. Intolerance of statin, due to moderate to severe myalgias and/or a 5-fold increase in creatinine kinase have also limited the beneficial use of optimised lipid therapy, and this statin-induced myopathy may represent up to 10 % of treated patients in a primary care setting.62 At present, PCSK9 inhibitors have successfully shown to significantly reduce LDL-C, non-HDL-C and Lp(a) in different patient population and clinical settings. As we look to continued phase III clinical trials, particularly those measuring CV outcomes, PCSK9 inhibitors offer promise to patients with significant residual risk that treatment with PCSK9 inhibitors can further lower cardiovascular outcome events and related mortality.