








Despite advances in therapy, heart failure (HF) continues to be a leading cause of hospitalisation in those over 65.1 Around 2 % of people in Europe live with HF and as many as 10 % of those over 75 years are affected.2 Prognosis continues to be poor, with approximately half of patients dying within 5 years of first hospitalisation,3 a more severe prognosis than many malignancies.4
Sleep-disordered breathing (SDB) may comprise obstructive sleep apnoea (OSA) or central sleep apnoea (CSA), although many patients have a mixed pattern that may change during the course of a night.5 SDB is common in HF with either reduced or preserved ejection fraction (EF) – around 50 % of patients are affected compared with less than 10 % of the general population.6,7
SDB is associated with an increased morbidity and mortality in patients with HF and there is some evidence that it is not merely a marker of poor prognosis, but may be a process with independent pathophysiological consequences that may accelerate the natural history of HF. It remains under-diagnosed and may be a therapeutic target in some patients.
This article reviews the aetiology and mechanisms of SDB and the current evidence for the investigation and management of this condition in patients with HF. The SERVE-HF randomised outcome trial8 has recently reported some unexpected results that have changed our view of CSA and this will be discussed.
Aetiology and Classification of Sleep-disordered Breathing in Heart Failure
SDB describes a range of conditions in which there is an abnormality of the breathing pattern during sleep. By definition, a cessation of oro-nasal airflow for over 10 seconds is termed an apnoea, while a reduction in airflow amplitude by 30 % or more for 10 seconds associated with a desaturation of 3 % or more is a hypopnoea.9 Events are further classified into obstructive, central or mixed depending on the presence, absence or characteristics of thoraco-abdominal movement during the episode. The number of events per hour of sleep is the apnoea–hypopnoea index (AHI). Up to 5 events per hour is considered normal, 5–15/hour mild, 15–30/hour moderate and over 30 events/hour severe SDB.9
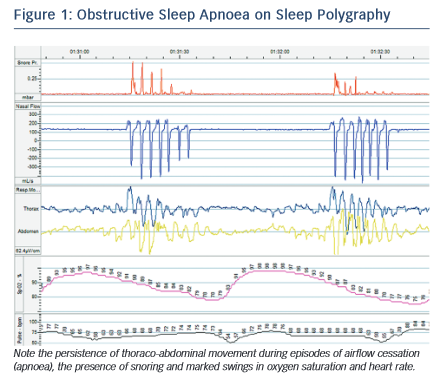
Although OSA and CSA have different underlying mechanisms, they are both driven by the internal pathophysiological environment found in HF. In OSA, the interruption of airflow is caused by collapse of the airway at the pharynx during inspiration. Pharyngeal muscle tone is decreased during sleep and, in HF, rostral shift of fluid from oedematous extremities to the pharynx may exacerbate this tendency. Research has demonstrated a significant increase in neck circumference overnight associated with a decrease in leg circumference in those with HF and OSA.10 Obesity and retrognaithism also predispose to this condition, although patients with HF and OSA are more frequently non- obese compared with patients with normal cardiac function.11 OSA is characterised by abrupt cessation of breathing as the airway occludes, accompanied by respiratory muscle effort and often snoring, followed by compensatory hyperventilation to restore arterial partial pressure of carbon dioxide (PaCO2) (see Figure 1).
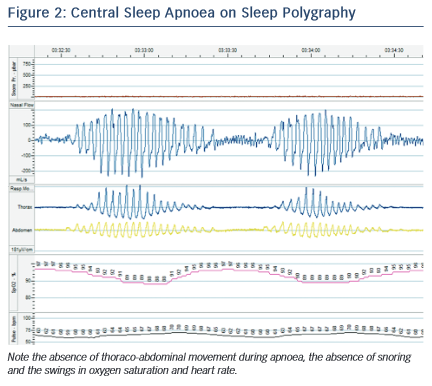
In CSA, the abnormal breathing pattern is mediated by dysregulation in the respiratory centres of the brainstem. While the mechanisms of CSA are not fully understood and may vary between individuals, the following processes are likely to contribute. In normal physiology, depth and frequency of breathing (and hence minute ventilation) is driven by PaCO2 sensed by the chemoreceptors in the carotid bodies, aortic arch and brainstem. Small rises in PaCO2 result in a transient increase in ventilation that restores the PaCO2 to within the narrow normal physiological range. In patients with CSA, there is an exaggerated hypercapnic ventilatory response, so that small rises in PaCO2 result in inappropriate hyperventilation, which drives the PaCO2 down. If the PaCO2 falls to below the ‘apnoeic threshold’ the neural drive becomes insufficient to stimulate a breath and an apnoea occurs. Lesser reductions in neural drive result in hypopnoea. The PaCO2 subsequently rises again and the cycle repeats.12 Exaggerated sympathetic nervous activity in HF is thought to underlie this excessive response and patients with CSA and HF have a low resting PaCO2.5 In addition, prolonged circulation time in HF results in a lag between the PaCO2 sensed in the brainstem and that at the alveoli, which exacerbates an overshoot of the feedback loop. Oedema and congestion in the lungs, which may progress during the night, stimulates pulmonary J receptors resulting in a reflex hyperventilation. A characteristic crescendo-decrescendo pattern of breathing in CSA is termed ‘Cheyne-Stokes respiration’ (see Figure 2).
Pathophysiological Effects of
Sleep-disordered Breathing
Although OSA is traditionally thought to accelerate cardiovascular deterioration, whereas CSA is predominantly a marker of severity of HF, both types of SDB share common deleterious pathophysiological consequences. However, there is evidence that CSA may in some cases be adaptive in HF.
In OSA, apnoea is accompanied by negative intrathoracic pressure as the respiratory muscles attempt to inspire against a collapsed airway. This enhances venous return to the right heart, increasing pre-load, which increases the work of the failing heart and pushes the septum to the left, further embarrassing LV function. In addition, negative intrathoracic pressure opposes contraction of the ventricular free wall, effectively increasing afterload. Recurrent episodes of apnoea and hypopnoea lead to arousals and sympathetic nervous system stimulation, known to be maladaptive in HF, and swings in heart rate and blood pressure.13 High concentrations of urinary catecholamines are found in patients with OSA14 and sleep quality is disrupted, often resulting in day-time somnolence and increased risk of road traffic accidents.15 In patients with OSA there is evidence of abnormal endothelial function – the vasodilatory response to nitric oxide is blunted and expression of the vasoconstrictors endothelin-1 and angiotensin II increased.14,16 OSA is associated with hypertension, coronary artery disease, stroke and arrhythmias, probably due to both shared risk factor profiles and the mechanisms described. These maladaptive mechanisms affect both systolic and diastolic cardiovascular function.
Perhaps unsurprisingly, patients with OSA and HF have a significantly increased mortality compared with those without OSA, particularly with co-existent ischaemic heart disease.17 In the Sleep Heart Health Study, a large community-based cohort observational study, the presence of severe OSA was associated with more than twice the all-cause mortality risk over 8 years of follow-up.18 OSA was also associated with a 58 % increase in risk of developing HF de novo.19 Patients with OSA and HF have an increased risk of sudden cardiac death, malignant arrhythmia and the need for implantable defibrillator therapy, particularly during the night; in patients with HF and either CSA or no SDB, malignant arrhythmias and defibrillator therapies occur predominantly during the day.20,21
CSA is also associated with a worsened prognosis in HF. In one study, the presence of any CSA (including mild disease) was associated with a significantly shorter mean survival in patients with EF <45 %: 45 months versus 90 months.22 This has traditionally been attributed to CSA being a marker of more severe cardiac dysfunction. However, there are reasons to believe that CSA itself may accelerate HF. Patients with CSA experience episodes of hypoxaemia and swings in heart rate and blood pressure, in a similar way to OSA (see Figure 2). CSA is associated with higher sympathetic nervous system activity, known to be maladaptive in HF,23 although there is debate as to whether this is due to the CSA itself or the underlying HF.24,25 A possible role for CSA in driving enhanced sympathetic activity is suggested by the finding that administered oxygen and continuous positive airway pressure (CPAP) both ameliorate sympathetic tone.26 The episodes of hyperventilation in CSA may also increase demands on the failing heart. However, in contrast to OSA, there is no marked negative intrathoracic pressure during apnoeas and hypopnoeas, as the respiratory muscles are not stimulated, so many of the haemodynamic effects of this (such as changes in ventricular pre- and after-load) are absent.
This view of CSA has been challenged recently by the unexpected findings of the SERVE-HF trial.8 In this randomised study, treatment of patients with HF and predominant CSA with adaptive servo- ventilation (a non-invasive ventilation technique known to ameliorate the peaks and troughs of ventilation in CSA very effectively) had no impact on the primary combined endpoint of time to death, life- saving cardiovascular intervention or unplanned HF hospitalisation, but did increase the risk of all-cause and cardiovascular mortality compared with controls (hazard ratio for death from any cause, 1.28, 95 % confidence interval [CI] 1.06–1.55; p=0.01). This surprising result raises the possibility that CSA may in fact be protective in HF and there are possible explanations for this, as described by Naughton in 2012.27 Indeed, in patients without cardiovascular disease, the Sleep Heart Health Study found that CSA was not associated with increased mortality.18 The increased mortality seen in patients with HF and CSA may be partly related to the difficulty in identifying an appropriate control group (as those with CSA tend to have more severe cardiac dysfunction even within the inclusion criteria of the studies).
There are several proposed mechanisms for the potential cardio- protective effects of CSA. The episodes of hyperventilation lead to end-tidal volume increases of 400 ml on average.28 This increases oxygen storage in the lung, reduces hypoxaemia in the presence of pulmonary oedema and impaired gas exchange and improves lung compliance in a similar way to CPAP therapy. The hyperventilation phase of CSA has been shown to reduce sympathetic and increase vagal tone, and the elevated sympathetic tone seen in those with HF and CSA relates more closely to the severity of HF than of CSA.25 Importantly, the episodes of hyperventilation induce a respiratory alkalosis. Hypocapnia has been shown to preserve myocardial function in the presence of hypoxia in dogs29 and alkalosis results in better myocardial performance during hypoxia in vitro.30 Hypocapnia and alkalosis also increases the oxygen- carrying capacity of haemoglobin, according to the Bohr and Haldane effects. As hypercapnia and acidosis are a frequent finding in acute decompensated HF, this may have a protective role. Swings in intrathoracic pressure with hyperventilation may also augment cardiac output via pump-like variations in pre- and after-load. In addition, hyperventilation is thought to reverse oedema-induced bronchoconstriction.31 During apnoeic episodes in CSA there is also slightly elevated intrathoracic pressure, which may prevent alveolar collapse.32 Furthermore, recurrent episodes of hypoxaemia may stimulate erythropoiesis and it is postulated that alternating high and low workload may reduce respiratory muscle fatigue and improve oxygenation compared with constant effort.33 The effect of adaptive servo-ventilation (ASV) on these protective mechanisms may, at least in part, explain the surprising increase in cardiovascular mortality found in the SERVE-HF trial.
Investigation of Sleep-disordered Breathing
Given the prevalence of SDB inpatients with HF, a high index of suspicion is appropriate. In the general population, questionnaires designed to screen for daytime somnolence, such as the Epworth sleepiness questionnaire are frequently used. However, patients with HF tend to report low daytime somnolence even in the presence of significant SDB,11 possibly due to increased sympathetic nervous activity. It is therefore usually necessary to take a confirmatory history from a partner where possible or to proceed to formal investigation where SDB is suspected. However, those with SDB do have increased measures of sleepiness when assessed objectively.34
Simple screening for SDB may be performed by overnight pulse oximetry. Using desaturations of ≥3 % at a cut off of 12.5 events/ hour for significant SDB, Ward et al. reported a sensitivity of 93 % and specificity of 77 % compared with formal polysomnography.35 With this approach, few patients with SDB would be missed, but pulse oximetry alone is unable to differentiate between OSA and CSA and further investigation is mandatory in patients with a high desaturation index or anyone in whom suspicion remains high despite a negative test.
Several non-contact bedside monitors are available which use ultra- low power radiowaves to detect respiratory movement during sleep. These can diagnose significant SDB with a sensitivity and specificity of around 90 %,36 but are not yet common in clinical practice.
The gold-standard test for SDB remains in-hospital polysomnography. This involves the patient spending a night in a monitored sleep laboratory while the equipment monitors the respiratory pattern, oxygen saturation, electrocardiogram, electroencephalogram and, in some cases, electromyogram and oculogram. This test provides unparalleled detail on the severity and type of SDB, as well as phases and architecture of sleep. It is, however, relatively expensive and laborious and therefore SDB is commonly diagnosed by more limited sleep polygraphy in the patient’s home. This test usually incorporates elastic effort straps around the chest and abdomen, a finger saturation probe and tubing under the nostrils to measure airflow. A thermistor may be included to estimate oral airflow and a snore sensor is also available. This is easily portable and can be fitted by the patient; it may provide more useful data in that sleep is less likely to be disrupted by the hospital environment and the data are relatively simple to interpret. Research has shown that polygraphy correlates well with polysomnography for the diagnosis of SDB in HF.37
Over the past 10 years, there has been interest in whether pacemaker respiratory sensors can be used to accurately diagnose and monitor SDB. Both simple and complex devices are now available that incorporate algorithms that use changes in transthoracic impedance with ventilation to give a respiratory disturbance index, akin to the AHI. In one study, a commercially available pacemaker algorithm showed a sensitivity of 88.9 % and a specificity of 84.6 % for the diagnosis of severe SDB.38 Evaluation of the accuracy of other available algorithms is currently underway (NCT02204865). At pacemaker download, up to 3 months of data on AHI are available and, with remote monitoring, changes in AHI could be a useful early indicator of HF decompensation. Further research is required.
Treatment of Sleep-disordered Breathing in Heart Failure
Optimal pharmacological therapy of HF, including careful attention to maintaining euvolaemia, would be expected to improve SDB of both sorts by reducing pharyngeal oedema, sympathetic drive and pulmonary congestion, although specific data are lacking. While OSA and CSA share many pathophysiological mechanisms and may co-exist in the same patient to varying degrees, the role of non- invasive ventilation (NIV) and some novel therapies are very different between the two.
Obstructive Sleep Apnoea
The approach to patients with OSA and HF includes advice regarding weight loss and sleep hygiene, including avoidance of excess alcohol or other sedatives as appropriate.39,40 This is relatively less useful in the HF population as OSA is less frequently associated with these factors than in the general population, but should be considered on an individual patient basis.
Non-invasive ventilation, particularly with CPAP, is well-established in current guidelines for non-HF patients with moderate to severe OSA and daytime somnolence (40). In this population, CPAP significantly improves the AHI and Epworth sleepiness score, but has not been consistently shown to improve hypertension, mortality or quality of life measures.40,41
The evidence for CPAP use in the HF population is based on fewer and smaller studies than the general population. In a randomised controlled trial of 24 patients with HF and OSA, Kaneko et al.42 found that 1 month of CPAP therapy improved left ventricular EF (LVEF) from 25.0 ± 2.8 % to 33.8 ± 2.4 % (p<0.001), reduced LV end systolic diameter from 54.5 ± 1.8 mm to 51.7 ± 1.2 mm (p=0.009) and reduced heart rate and blood pressure in parallel with a significant decrease in AHI (37.1 ± 6.4/hour to 8.3 ± 2.8/hour; p<0.001). There was no significant change in these parameters in the control group. Mansfield et al. randomised 55 patients with HF and OSA to CPAP for 3 months or control. Those receiving CPAP had a greater improvement in LVEF (5.0 ± 1.0 % versus 1.5 ± 1.4 %; p=0.04), reduced urinary noradrenaline and improved quality of life scores.43 In a larger non-randomised trial, Kasai et al. followed 88 patients with HF and moderate to severe OSA for around 2 years.44 They demonstrated that those not treated with CPAP had a significantly higher risk of death or hospitalisation (HR 2.03, 95 % CI 1.07–3.68; p=0.030) than those receiving CPAP. Those with poor CPAP compliance also had significantly worse outcomes. Physiologically, OSA has been shown to acutely reduce stroke volume and cardiac output overnight and that this reduction is ameliorated by CPAP therapy.45 Measures of cardiac sympathetic tone are also reduced by CPAP in those with OSA.46
There are conflicting reports regarding the impact of cardiac resynchronisation therapy (CRT) on OSA.47,48 The majority of research has demonstrated no significant improvement in AHI with CRT, in contrast to those with CSA. In some patients with retrognathism or tongue enlargement, mandibular advancement devices have been shown to be effective.49 Hypoglossal nerve stimulation is a novel technique aimed at maintaining pharyngeal tone during sleep. A non-controlled study of 126 general OSA patients demonstrated a 68 % reduction in AHI with this technique, but the role in patients with HF is not known.50
Central Sleep Apnoea
Following successful trials of CPAP in OSA and HF, the CANPAP trial sought to evaluate its use in those with CSA.51 This study randomised 258 patients with HF and CSA to CPAP or optimal medical therapy only with a mean follow up of 2 years. CPAP was effective at improving AHI (–21 ± 16 versus –2 ± 18 per hour; p<0.001) and nocturnal oxygen saturation, as well as plasma noradrenaline concentration, LVEF and 6-minute walk distance. However, CPAP had no overall influence on rates of hospitalisation and death compared with controls. A post- hoc analysis did demonstrate a survival advantage in those treated with CPAP in whom AHI was supressed to below 15/hour, raising the possibility that more efficacious ventilatory techniques may provide survival benefit to the wider population with HF and CSA.52
A more complex non-invasive ventilatory modality is ASV. In this mode, the ventilator automatically adjusts the degree of respiratory support such that more inspiratory positive airway pressure (IPAP) is provided during episodes of hypopnoea and apnoea and less during hyperpnoea. It also provides positive end expiratory pressure (PEEP) as in CPAP and mandatory breaths during apnoeas. It is therefore potentially effective in both CSA and OSA.
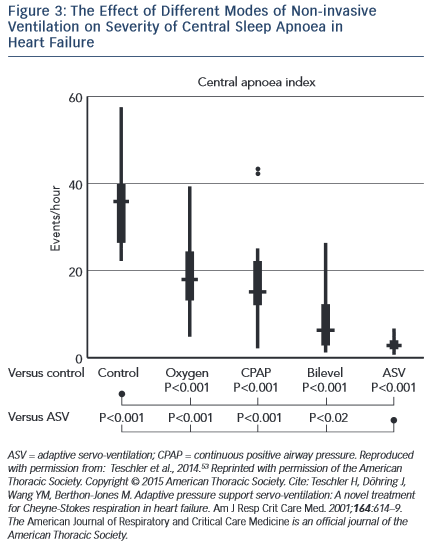
Several studies have demonstrated benefits of ASV on surrogate endpoints in CSA and HF. Teschler and colleagues showed that ASV was significantly more efficacious at suppressing CSA than CPAP, bi-level positive airway pressure (BiPAP) or oxygen therapy (see Figure 3).53 In an observational study, Oldenburg and colleagues found that 6 months of ASV therapy in those with HF and CSA almost eradicated CSA, from 37.4 ± 9.4/h to 3.9 ± 4.1/h (p=0.001) and was associated with markedly improved cardiopulmonary exercise test parameters, increased LVEF (28.2 ± 7 % to 35.2 ± 11 %; p=0.001) and reduced N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) concentration.54 These results were largely replicated by Hastings et al.55 Similar improvements were found in those with HF with preserved EF (HFPEF).56 Reductions in sympathetic nervous activity have also been demonstrated with ASV therapy.57 Furthermore, registry data have revealed a significantly longer time to first implantable cardioverter defibrillator (ICD) therapy or ventricular arrhythmia in patients with HF, CSA and an ICD treated with ASV compared with those not treated58. Meta-analysis of trials suggested an overall reduction in AHI, improved exercise capacity, LVEF and quality of life measures with ASV treatment compared with controls.59 However, these trials were not powered to detect changes in harder clinical endpoints.
SERVE-HF is the first large-scale and longer-term randomised trial to report the effect of ASV on clinical outcomes.8 This multi-centre trial enrolled 1,325 patients who were randomised to ASV in addition to optimal medical therapy or optimal medical therapy only for a median of 31 months. The trial was powered for the combined endpoint of all-cause death, unplanned hospitalisation for worsening HF, heart transplantation, cardiac arrest or appropriate ICD therapy. As discussed above, ASV did not influence the primary end-point but was associated with a significant increase in both all-cause and cardiovascular mortality. Most of the excess mortality related to sudden death, presumably from cardiac arrhythmia, and not only during the night. Based on this study, the largest of its kind in HF and SDB, and despite the beneficial effects demonstrated previously, ASV cannot be recommended for those with CSA and HF.
In the randomised CHF-HOT study of 97 patients with CSA and HF, overnight oxygen therapy reduced AHI and improved LVEF and New York Heart Association (NYHA) grades,60 but the effect on clinical outcomes is unknown. Inhaled carbon dioxide is also effective at reducing AHI, but not arousals or sympathetic nervous activity.61
CRT produces a significant reduction in AHI in appropriately selected patients,62 presumably due to improved cardiac output, reduced pulmonary congestion and ameliorated sympathetic tone. Research is underway to determine whether improvement in CSA with CRT is associated with normalisation of the hypercapnic ventilatory response (NCT02203383). It is unknown whether the presence of CSA should be a factor when considering a patient for CRT.
A novel therapy for CSA is phrenic nerve stimulation. This involves the implantation of a pacemaker-like device with a lead that communicates with the phrenic nerve via the left pericardiophrenic or right brachiocephalic vein. The device stimulates the phrenic nerve when no intrinsic impulse is detected for a specified time period and this stimulates a breath, preventing the hypercapnia, which triggers the cycles of Cheyne-Stokes respiration. Early research suggests a significant improvement in AHI, oxygenation and arousals, but the impact on prognosis is unknown.63 Further research is underway.
Conclusion
SDB in HF is common, frequently undiagnosed and associated with a poor prognosis. The use of sleepiness questionnaires is not useful in HF. While polysomnography remains the gold standard test, simple screening may be performed with overnight pulse oximetry. Home polygraphy is simple to use and interpret with adequate diagnostic power in most patients. In parallel with optimal HF treatment, therapy with CPAP is likely to be of benefit for those with HF and OSA. The role of positive pressure support in CSA is unclear. ASV has many advantages in CSA and HF but this does not appear to translate in to improved clinical endpoints and it increases mortality. The long- term results of novel therapies such as phrenic nerve stimulation are awaited. Our understanding of SDB continues to evolve rapidly and there are likely to be major changes in our approach to the condition in the coming years.