










In recent decades, structural and functional abnormalities of the coronary microvasculature, referred to as coronary microvascular dysfunction (CMD), have been implicated across the spectrum of cardiovascular diseases.1,2 The true prognostic impact of CMD in patients with angina but without obstructive coronary artery disease (CAD) is still debated, largely due to the heterogeneity of previous studies, depending on the presence or absence of concomitant non-significant CAD (<50% stenosis), in which acute coronary events (e.g. plaque rupture) often occur.2 Accordingly, a meta-analysis revealed that the long-term prognosis of this population was heterogeneous and depended on the presence of ischaemia, as verified by non-invasive imaging techniques (stress echocardiography or nuclear imaging), which was associated with a higher incidence of worse clinical outcomes.3
However, CMD has attracted much attention in view of its significant prognostic impacts in various cardiovascular diseases.4,5 The prevalence of CMD in cardiovascular disease is not negligible, and a large cohort study (n=1,439) reported that two-thirds of patients with chest pain but without obstructive CAD had endothelium-dependent or -independent CMD.6 Moreover, 42% of patients with chest pain who presented to an emergency department and in whom acute MI was ruled out, as well as approximately 10% of patients with acute MI, had CMD.7,8 A brief comparison of contemporary guidelines for the diagnosis and management of patients with microvascular angina and CMD is presented in Table 1.9–11
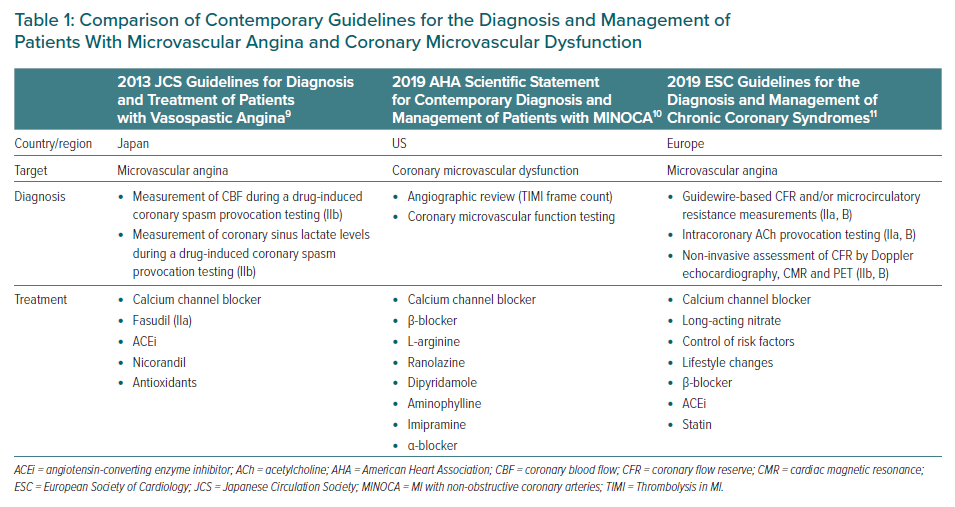
Chronic low-grade vascular inflammation plays an important role in the mechanisms underlying CMD, especially in patients with diabetes, obesity, chronic inflammatory rheumatoid diseases and heart failure with preserved ejection fraction (HFpEF).5,12 The endothelium plays a pivotal role in modulating vascular tone by synthesising and releasing endothelium-derived relaxing factors (EDRFs), including vasodilator prostaglandins, nitric oxide (NO) and endothelium-dependent hyperpolarisation (EDH) factors, in a distinct vessel size-dependent manner. NO predominantly mediates vasodilatation of relatively large, conduit vessels (e.g. the aorta and epicardial coronary arteries), whereas EDH factors mediate vasodilatation in small resistance vessels (e.g. arterioles and coronary microvessels; Figure 1).13,14 Inflammatory processes in the vascular wall can be accompanied by endothelial dysfunction, characterised by a reduction in the production and/or action of EDRFs, instigating the first step towards coronary atherosclerosis.14 A recent meta-analysis found a positive association between higher serum interleukin (IL)-1 receptor antagonist concentrations and an increased incidence of cardiovascular disease in the general population.15 Moreover, positive results of the recent randomised controlled trials targeting inflammatory mediators further support the inflammatory hypothesis of atherosclerotic cardiovascular diseases.16–18 In this review, we provide a concise overview of the current knowledge of the involvement of inflammation in the pathophysiology and molecular mechanisms of CMD from bench to bedside.
Pathophysiology of Coronary Microvascular Dysfunction
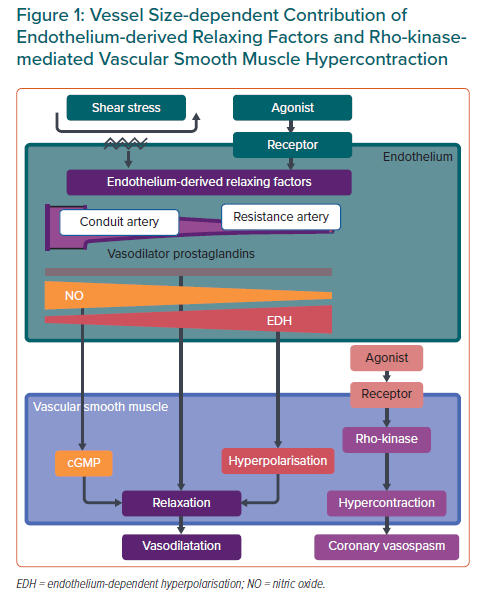
There are three central mechanisms underlying coronary vasomotion abnormalities: enhanced coronary vasoconstrictive reactivity (e.g. coronary spasm) at the epicardial and microvascular levels, impaired endothelium-dependent and -independent coronary vasodilator capacity and increased coronary microvascular resistance induced by structural factors (e.g. luminal narrowing, vascular remodelling, vascular rarefaction and extramural compression; Figure 2). The underlying mechanisms of CMD may be heterogeneous (including several structural and functional alterations) often coexist in various combinations and can cause myocardial ischaemia even in the absence of obstructive CAD.6,19
The major mechanism in the pathogenesis of coronary artery spasm is Rho-kinase-induced myosin light chain phosphorylation with resultant vascular smooth muscle cell (VSMC) hypercontraction (Figure 1).13,20,21 Intracoronary administration of the Rho-kinase inhibitor fasudil is effective in relieving not only refractory epicardial coronary spasm resistant to nitrates or calcium-channel blockers, but also coronary microvascular spasm and percutaneous coronary intervention (PCI)-related refractory myocardial ischaemia.22–24 However, fasudil has not been approved for clinical use by the European Medicines Agency or the US Food and Drug Administration.
We recently demonstrated that fasudil-induced decreases in microvascular resistance (IMR) were greater in patients with vasospastic angina (VSA) who had higher IMR values.19 This evidence supports the role of Rho-kinase activation in increased coronary microvascular resistance. In addition, enhanced epicardial and coronary microvascular spasms are associated with the increased production of mediators of vasoconstriction (e.g. serotonin), as well as inflammatory conditions (see below).25
Along with VSMC dysfunction, such as coronary artery spasm, endothelial dysfunction serves as another major mechanism involved in the pathogenesis of CMD. Inflammatory conditions and several key proinflammatory cytokines are also involved in the development of endothelial dysfunction.26 It is widely accepted that EDH factors, rather than NO, predominantly mediate the endothelium-dependent vasodilatation of resistance arteries.13,26 Thus, EDH factor-mediated vasodilatation is an important mechanism, especially in the microcirculation, where blood pressure and organ perfusion are finely tuned to meet fluctuating demand in the body (Figure 1). Comprehensive reviews of the detailed mechanisms of endothelial modulation of vascular tone are available elsewhere.13,14,26
Inflammation, Perivascular Adipose Tissue and Vasospastic Angina
Previous studies by us and others have revealed close relationships among inflammation, perivascular adipose tissue (PVAT) and vasa vasorum in the pathogenesis of coronary vasomotion abnormalities (Figure 2), as summarised in recent review articles in this Journal.27,28 For example, in vivo, the major inflammatory cytokine IL-1β induced intimal thickening and coronary vasospastic responses to intracoronary serotonin or histamine via ‘outside-to-inside’ signalling in pigs.29 In addition, in CAD patients who underwent elective coronary artery bypass grafting, the proinflammatory properties of the epicardial adipose tissue were significantly greater than those of subcutaneous adipose tissue, but this was not reflected by plasma concentrations of systemic inflammatory biomarkers or attenuated by chronic treatment with statins or angiotensin-converting enzyme inhibitors/angiotensin II receptor blockers.30
Using multimodality imaging techniques, including micro-CT and optical frequency domain imaging, we previously demonstrated that enhanced adventitial vasa vasorum formation was associated with coronary hyperconstriction via Rho-kinase activation, a predominant mechanism of coronary vasospasm, in patients with VSA.27,31 The vasa vasorum serves as a conduit for inflammatory cells and cytokines originating from the surrounding inflamed adipose tissue to the local coronary atherosclerotic lesions in the vascular wall (Figure 2).
The novel link between inflammation and coronary vasomotion abnormalities is further supported by the findings that coronary vasoconstriction in response to acetylcholine (ACh) in patients with early CAD was greater in coronary artery segments with than without macrophage infiltration and vasa vasorum proliferation in an additive manner, indicating an important role of inflammation and vasa vasorum proliferation in the pathogenesis of CAD.32 It is conceivable that Rho-kinase activation and inflammatory responses are the shared mechanisms underlying enhanced coronary vasoconstrictor reactivity (e.g. coronary spasm) at both the epicardial and microvascular levels, and thus similar phenomena will be seen in the coronary microvasculature as in the epicardial coronary arteries.
There is growing evidence of a role for PVAT in the regulation of vascular tone in a wide variety of experimental and clinical settings, adding another layer of complexity to PVAT-mediated responses.33 PVAT has different pathophysiological roles depending on its location in the body and modulates vascular tone in a paracrine/autocrine manner by releasing an array of vasoactive mediators, including adiponectin, NO, hydrogen sulfide and others yet to be identified.33 For example, interscapular brown adipose tissue exerts an anticontractile effect through hydrogen peroxide (H2O2)-induced cGMP-dependent protein kinase G (PKG) 1α activation and subsequent vasodilatation of small resistance arteries in mice.34 Considering that this oxidant-mediated PKG1α activation is a shared vasodilator mechanism of H2O2 as an EDH factor in coronary and mesenteric resistance arteries, targeting brown adipose tissue may be of therapeutic potential for the treatment of CMD.35–37 Perivascular inflammation has been shown to be associated with enhanced coronary vasoconstrictor reactivity in patients with VSA (Figure 2).28,38 Coronary PVAT volume measured by CT coronary angiography at the spastic coronary segment was significantly increased and positively correlated with the extent of coronary vasoconstriction provoked by intracoronary ACh in patients with VSA, although the total volume of epicardial adipose tissue was unchanged.28,38 In addition, coronary adventitial and PVAT inflammation evaluated by 18F-fluorodeoxyglucose PET/CT was significantly increased in patients with VSA, along with enhanced adventitial vasa vasorum formation and Rho-kinase activity of circulating leucocytes.38 Of note, the extent of coronary perivascular inflammation was markedly decreased in the spastic coronary artery after treatment with calcium channel blockers, which are the mainstay for the treatment of VSA.38,39
Sex Differences in Inflammation and Coronary Microvascular Dysfunction
Accumulating evidence indicates distinct sex differences in endothelial function and coronary vasomotion abnormalities, with major clinical implications.40,41 For example, the findings of the WISE study provide valuable insights into the aetiology of myocardial ischemia in female patients with chest pain in the absence of obstructive CAD, which is commonly attributed to CMD.42,43 Given the lower prevalence of obstructive CAD and higher prevalence of CMD in female patients in this population, the proper assessment and diagnosis of coronary functional rather than structural abnormalities should be considered.6
Many mechanisms have been proposed for the sex differences in the characteristics of CMD, including differences in sex hormone effects, autonomic regulation, genetic polymorphisms and susceptibility to proatherogenic mediators, such as inflammation, oxidative stress, endothelin-1 and angiotensin II.44,45 For example, oestrogens exert protective effects on endothelium-dependent vasodilatation through an anti-inflammatory effect on endothelium-derived mediators.26 In-depth reviews on this topic are available elsewhere.40,41
Inflammatory Modulators of Coronary Vascular Function: Beyond a Marker?
High-Sensitivity C-reactive Protein
Low-grade inflammation, such as that associated with elevations in high-sensitivity C-reactive protein (hs-CRP), significantly affects morbidity in atherosclerotic cardiovascular diseases and hs-CRP, among others, is an easily obtainable practical marker to identify patients at risk of long-term mortality in many clinical settings. In a large-scale prospective cohort study of postmenopausal women with no history of cardiovascular disease or cancer, increased plasma markers of inflammation at baseline, such as hs-CRP, serum amyloid A, IL-6 and soluble intercellular adhesion molecule type 1 (sICAM-1), were significantly associated with the risk of future cardiovascular events.46 Of these markers, hs-CRP was the strongest predictor of the risk, and the addition of hs-CRP measurement to standard lipid screening better identified women at increased risk.46
In another study, sympathetic stimulation (cold pressor testing)-induced changes in epicardial luminal area and myocardial blood flow responses were significantly impaired in patients with elevated serum CRP concentrations (≥0.5 mg/dl) who had coronary risk factors but angiographically normal coronary arteries.47 Similarly, in patients with chest pain but angiographically normal coronary arteries, elevated CRP levels were associated with impaired coronary blood flow (CBF) responses to intracoronary ACh, but not with the change in coronary artery diameter (conduit vessels), suggesting a possible relationship between chronic vascular inflammation and CMD.48 In patients with typical angina and transient myocardial ischaemia despite angiographically normal coronary arteries, elevated hs-CRP concentrations (>0.3 mg/dl) were associated with CMD, as assessed by PET-derived myocardial blood flow and coronary flow reserve (CFR).49 This study provides the first direct evidence for a relationship between low-grade chronic inflammation and CMD.49
In patients with typical angina but without CAD, serum concentrations of soluble CD40 ligand and tumour necrosis factor (TNF)-α were significantly associated with a decrease in the myocardial perfusion reserve index (MPRI) during adenosine stress cardiac magnetic resonance (CMR), whereas serum concentrations of TNF-α and sICAM-1 were significantly associated with reductions in MPRI during ACh stress CMR.50 Reduced MPRI was significantly correlated with both endothelium-dependent and -independent changes in CBF in response to ACh and adenosine, respectively.50
Patients with acute chest pain who were found to have CMD, as assessed by reduced PET-derived CFR, but no evidence of CAD had elevated levels of serum renalase, an anti-inflammatory protein released from the heart and kidneys in response to acute ischaemic stress.51 Of note, these patients had similar concentrations of serum inflammatory markers (CRP, TNF-α, vascular endothelial growth factor, IL-1β, IL-6 and metalloproteinases) as control patients.51 These findings imply the dynamic nature of the inflammatory response to an acute ischaemic insult compared with chronic coronary syndrome.51
Another recent study using encompassing proteomic analysis of cardiovascular disease biomarkers also confirmed that the proinflammatory IL-1β/TNF-α/IL-6/CRP pathway was significantly associated with CMD in women with angina but no obstructive CAD.52 Together, these observations suggest that chronic subclinical inflammation contributes to the development of CMD.
Soluble Urokinase-Type Plasminogen Activator Receptor
Among several inflammatory mediators implicated in the pathogenesis of CMD and atherosclerotic cardiovascular diseases, soluble urokinase-type plasminogen activator receptor (suPAR) has emerged as an inflammatory marker with atherogenic properties and a potential contributor to endothelial dysfunction.53 Recent studies have unveiled an association between suPAR and CMD in patients with non-obstructive CAD.54 The net production of suPAR across the coronary circulation can be calculated as follows:
suPAR = (coronary sinus concentration − left coronary arterial concentration) × basal CBF
Positive values indicate the production or release of suPAR, whereas negative values indicate the retention or metabolism of suPAR in the coronary circulation.54 Patients with endothelium-dependent CMD showed net transcoronary production of suPAR.54
There are three possible explanations for the link between endothelium-dependent CMD and epicardial coronary atherosclerosis based on the important role of suPAR in the pathogenesis of endothelium-dependent CMD and coronary atherosclerosis. First, the transcoronary production of suPAR in patients with endothelium-dependent CMD may reflect a local low-grade inflammatory state in the coronary circulation, a key process underlying atherosclerosis and endothelial dysfunction.26 This is in agreement with prior observations showing enhanced transcoronary production of inflammatory cytokines, such as lipoprotein-associated phospholipase A2 and IL-8, in patients with endothelium-dependent CMD.55
Second, suPAR may be an active promoter of endothelium-dependent CMD, based on early evidence showing suPAR-mediated cleavage and inactivation of vasodilator peptides such as calcitonin gene-related peptide, as well as the generation of vasoconstrictor endothelin-1.56 Third, suPAR may promote the development and progression of coronary atherosclerosis in a distinct milieu exposed to endothelium-dependent CMD. suPAR has been shown to be involved in the pathogenesis of atherosclerosis through macrophage foam cell formation, the migration and proliferation of VSMCs and the formation of vulnerable plaques.53 The polarised release of suPAR from the inflamed vascular wall in the basolateral direction acts locally within the plaque to accelerate atherosclerosis, whereas circulating suPAR reflects only a fraction of the receptor pool, which is shed from various sources in the body.57
These observations may provide insights into the mechanisms by which endothelium-dependent CMD, likely in association with altered patterns of endothelial shear stress or suPAR-mediated inflammatory processes, contributes to the development of epicardial coronary atherosclerosis, even though these focal lesions are located upstream to the microcirculation.58,59
Hallmarks of Disease
Obesity
Reduced CFR in obese patients without obstructive CAD is associated with increased plasma IL-6 and TNF-α concentrations, independent of cardiovascular risk factors.60 These proinflammatory adipokines may contribute to the evolution and progression of CMD by virtue of their ability to induce vascular inflammation and disrupt microvascular systems. For example, aging and obesity induced a phenotype transition of PVAT into a proinflammatory state by increasing the activity of a disintegrin and metalloproteinase 17 (ADAM17) activity and soluble TNF release in adipose tissue, leading to impaired bradykinin-induced endothelium-dependent vasodilatation of human coronary arterioles and thereby the development of CMD.61 Moreover, coronary PVAT from obese pigs augmented the constriction of isolated coronary arteries in pigs, which occurred independent of coronary endothelial function.62 These observations implicate potential roles of PVAT-derived factors in the pathogenesis of obesity-related CMD.
Obesity also impairs PVAT-mediated vascular function through several mechanisms involving EDRFs. First, obesity has been shown to promote the recruitment of proinflammatory macrophages to PVAT and to impair the vasodilator properties of PVAT by reducing endothelial and VSMC production of hydrogen sulfide, a potent gaseous relaxing factor, leading to microvascular endothelial dysfunction.63 Second, in obesity, hypoxic conditions in expanding obese adipose tissue increased the expression of hypoxia-inducible factor-1α, which induced adipose tissue fibrosis and local inflammation, resulting in the overproduction of leptin, resistin, IL-6 and TNF-α.64 These proinflammatory adipokines and cytokines reach the coronary microcirculation via ‘inside-to-outside’ signalling to cause their remote effects by increasing oxidative stress in the coronary arteriolar wall, leading to reduced bioavailability of NO and impaired vasodilatation.64 Together, these lines of evidence indicate the therapeutic potential of targeting PVAT in the treatment of CMD.
In addition, obesity has been acknowledged as a distinct phenotype of HFpEF, characterised by structural, functional and haemodynamic alterations in the heart in favour of impaired exercise capacity, higher biventricular filling pressure in response to exercise and reduced pulmonary vasodilator reserve.65 The emerging interactions among systemic inflammation, CMD and HFpEF are discussed below.
Heart Failure With Preserved Ejection Fraction
HFpEF is a common and globally recognised form of heart failure that accounts for approximately 50% of cases. A chain of events driven by systemic inflammation has been proposed as a new paradigm for the pathogenesis of HFpEF.66 Briefly, comorbidities that are common in patients with HFpEF (e.g. diabetes, hypertension and obesity) can elicit a systemic proinflammatory state. Elevated levels of inflammatory mediators, such as IL-6, TNF-α, soluble suppression of tumorigenicity 2 (ST2) and pentraxin 3, provoke coronary microvascular endothelial inflammation and promote the production of reactive oxygen species (ROS), leading to reductions in NO bioavailability, cGMP content and PKG activity in adjacent cardiomyocytes, which favours the development of hypertrophy and increases resting tension because of hypophosphorylation of titin.67,68 Consequently, stiff cardiomyocytes and interstitial fibrosis contribute to high diastolic left ventricular stiffness and the development of heart failure. Such paracrine endocardial–myocardial interactions may explain how an inflamed coronary microvasculature can modulate myocardial structure and function, and thereby affect the trajectory of the disorder.
Accumulating evidence highlights the high prevalence and pathophysiological relevance of CMD in patients with HFpEF.1,69,70 A pioneering study showed that ACh-induced endothelium-dependent increases in CBF were impaired in heart failure patients with isolated left ventricular diastolic dysfunction, suggesting that coronary endothelial dysfunction may be an underlying mechanism of diastolic dysfunction.71 More recently, a relatively large cohort study of patients with HFpEF demonstrated a comparably high prevalence of both endothelium-dependent and -independent CMD, assessed by gold-standard invasive coronary reactivity testing.72 Intriguingly, endothelium-independent CMD was associated with worse diastolic dysfunction and increased mortality, although the measurements of diastolic function and haemodynamics were performed non-invasively only at rest and the effect of endothelium-dependent CMD may have been underestimated in that study.72 Indeed, a subsequent study from the same group demonstrated that pulmonary artery wedge pressure in HFpEF patients with endothelium-dependent CMD was only significantly elevated during exercise, and not at rest.73
Moreover, significant associations among endothelium-independent CMD, many inflammatory biomarkers and cardiac diastolic dysfunction were noted in women with angina but no flow-limiting coronary artery stenosis, further supporting the role of systemic inflammation in both CMD and HFpEF, as well as providing insights into potential mechanisms underlying the pathophysiology of HFpEF.74 In this regard, a comprehensive invasive assessment of coronary endothelial function by ‘functional coronary angiography’ is safe, feasible and of diagnostic value to better differentiate between endothelium-dependent and -independent CMD.
In light of the fact that H2O2 has potent vasodilator properties in coronary resistance vessels where EDH factor-mediated responses become relatively dominant to NO-mediated relaxations, it is conceivable that impaired H2O2/EDH factor-mediated vasodilatation may be involved in the pathogenesis of HFpEF driven by CMD. Indeed, we have recently demonstrated that CMD mediated by reduced H2O2/EDH factor-mediated vasodilatation is associated with cardiac diastolic dysfunction in mice lacking endothelial NO synthase.75
In line with these observations, a subanalysis of the PROMIS-HFpEF study showed that CMD was highly prevalent (75%) in patients with HFpEF and independently associated with increased risk of cardiovascular death and recurrent heart failure hospitalisation after adjusting for age, sex and confounding comorbidities.5
These studies suggest that CMD may be a promising therapeutic target for patients with HFpEF. Although no established treatment for CMD is available as yet in clinical practice, a recent animal study showed that treatment with a histone acetyltransferase p300 inhibitor in surtuin-3-knockout mice improved CMD in association with reductions in the expression of proinflammatory mediators, such as nuclear factor (NF)-κ and vascular cell adhesion molecule 1, in the coronary artery.76 Of note, contrary to the expectation that augmenting NO-mediated vasodilatation could benefit patients with HFpEF, the results of randomised clinical trials of the systemic and long-term administration of inorganic nitrite, used as an NO donor, in HFpEF patients were discouraging, or even harmful.77 These lines of evidence indicate that it is important to avoid excessive NO supplementation.78 Nitrosative stress induced by an excessive amount of NO may explain this ‘paradox’ of NO-targeted therapy, implicating the importance of a physiological balance between NO and EDH factors in endothelium-dependent vasodilatation.79,80
Chronic Inflammatory Rheumatoid Diseases
In patients with chronic inflammatory rheumatoid diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and systemic sclerosis (SSc), coronary microvascular structure and function can be affected in a chronic inflammatory milieu, leading to the development of myocardial ischaemia and increased cardiovascular morbidity and mortality, even in the absence of obstructive epicardial CAD.12 Indeed, patients with such autoimmune rheumatic diseases are characterised by accelerated atherosclerosis and premature CAD, and are often found to have CMD in a subclinical, asymptomatic manner.12 A high prevalence of CMD defined by reduced CFR was unexpectedly found in patients with SSc in the absence of clinical evidence of ischaemic heart disease.81 Moreover, a marked reduction in CFR (<2.0) was noted in the diffuse form of SSc.81 Similarly, CFR calculated as PET-derived adenosine/resting myocardial blood flow was reduced in patients with RA and SLE, even in the absence of both epicardial coronary stenoses and conventional coronary risk factors.12
Another study also noted that the presence of a chronic inflammatory disease, such as RA, SLE or SSc, was an independent predictor of CMD even in the absence of obstructive CAD.82 However, in that study, the CT-derived coronary artery calcium score in the left anterior descending coronary artery was not associated with CMD.82 It is possible that prolonged systemic inflammation may precede and contribute to the development of premature CAD in patients with a chronic inflammatory disease. This is supported by the finding that high IL-6 levels in patients with RA are accompanied by elevated concentrations of circulating asymmetrical dimethylarginine and endothelial dysfunction, manifested as impaired flow-mediated dilatation, despite the absence of atherosclerosis.83
Psoriasis is another common chronic inflammatory skin disease. Psoriasis is characterised by systemic inflammation affecting multiple organs in the body, including the cardiovascular system, and is associated with an increased risk of cardiovascular disease.84 In patients with psoriasis without overt CAD, a high prevalence of CMD, defined as PET-derived myocardial flow reserve <2.0, has been found, regardless of conventional coronary risk factors or the burden of coronary atherosclerosis.84 Patients with psoriasis treated with TNF inhibitors showed marked improvement in CFR in parallel with reductions in serum hs-CRP and TNF-α concentrations, further supporting the role of systemic inflammation in the mechanisms underlying CMD in patients with psoriasis.85 Considering the high prevalence and prognostic significance of CMD in patients with psoriasis, early recognition of the underlying CMD is indispensable in optimising the therapeutic strategy for these patients.
Inflammatory endothelial activation and oxidative stress may be a potential mechanistic link between chronic inflammatory rheumatoid diseases and CMD. For example, in a mouse model of RA, endothelium-dependent relaxation in response to ACh was impaired and inversely correlated with serum monocyte chemotactic protein-1 (MCP-1) concentrations, one of the major proinflammatory chemokines involved in the pathogenesis of RA and atherosclerosis.86 This endothelial dysfunction was ameliorated by statin therapy through inhibition of NF-κB binding to the MCP-1-induced protein gene enhancer.86
Therapeutic Potential of Anti-inflammatory Drugs
Non-steroidal Anti-inflammatory Drugs
Although low-dose aspirin is the mainstay of secondary prevention of atherosclerotic cardiovascular diseases, a higher dose is needed to provide a systemic anti-inflammatory effect.87 In a small, randomised trial of high-dose salsalate for 4 weeks, endothelium-dependent vasodilatation was unexpectedly impaired by the treatment, suggesting possible unfavourable effects of anti-inflammatory doses of salsalate on endothelial function.87 This is explained, in part, by the fact that vasodilator prostaglandins play a small but constant role independent of vessel size in general, but their contribution is not negligible in the kidneys; prostaglandin E2 and prostacyclin play important roles in the regulation of glomerular filtration rate, renal blood flow and renovascular tone.26
Canakinumab
Based on the inflammatory hypothesis of atherosclerotic cardiovascular diseases, a large-scale randomised clinical trial, namely the CANTOS study, was performed to examine whether targeting IL-1β could reduce the risk of recurrent cardiovascular events in patients with a history of myocardial infarction who had an hs-CRP level ≥0.2 mg/dl.16 Notably, the study population had a persistent proinflammatory response despite the use of aggressive secondary prevention strategies.16 Treatment with canakinumab, a selective anti-IL-1β monoclonal antibody, at a dose of 150 mg every 3 months significantly reduced the rate of the primary efficacy end point, which was a composite of non-fatal MI, non-fatal stroke or cardiovascular death, compared with placebo.16
Moreover, subanalysis of CANTOS showed that, independent of LDL levels, the magnitude of the reduction in hs-CRP was strongly associated with the reductions in cardiovascular events and all-cause mortality following canakinumab therapy; post-MI patients who achieved hs-CRP concentrations <0.2 mg/dl following a single dose of canakinumab had a 25% reduction in major adverse cardiac events and a 31% reduction in all-cause mortality compared with those who achieved hs-CRP concentrations ≥0.2 mg/dl.88 The results of CANTOS build on the inflammatory hypothesis of atherothrombosis, suggesting that targeting IL-1β (which drives the IL-6-mediated inflammatory pathways), rather than non-specific elimination of inflammatory mediators, may be beneficial in patients who have a residual proinflammatory response.
Colchicine
In recent years, the classical anti-inflammatory agent colchicine has attracted renewed attention for the treatment and secondary prevention of cardiovascular diseases. Two landmark clinical trials, namely the COLCOT and the LoDoCo2 trials, have demonstrated the efficacy and safety of low-dose colchicine at a dose of 0.5 mg/day in patients with stable CAD.17,18 A subanalysis of COLCOT revealed that a significant reduction in the primary endpoint was evident in patients who were initiated on colchicine within the first 3 days after MI, but not in those who were treated thereafter.89 In line with these results, the administration of low-dose colchicine at the same dose for 1 week significantly reduced the serum hs-CRP concentrations and improved endothelial function evaluated by flow-mediated dilatation in patients with CAD whose white blood cell count was ≥7,500 /µl.90 Although colchicine may have therapeutic potential in patients with CMD associated with inflammation, it should be noted that some studies reported a greater occurrence of non-cardiovascular deaths in the colchicine-treated groups.18,91
Conclusion
Patients with coronary vasomotion abnormalities are often complicated by inflammatory responses and peripheral endothelial dysfunction, in which CMD manifests as systemic vascular dysfunction beyond the heart.92 This further supports the concept of ‘primary coronary microcirculatory dysfunction’, and has important implications for practice and research.93
The results of two recent landmark clinical trials regarding the management of stable CAD, namely the ORBITA trial and the ISCHEMIA trial, have questioned the benefit of PCI and suggest the importance of coronary microvascular physiology.94,95 Although these trials did not focus directly on coronary microvascular function, an interesting speculation is that CMD may contribute to cardiac ischaemia even after successful revascularisation of significant epicardial coronary stenosis.
Whether the management of CMD by targeting inflammatory mediators can improve the outcome of patients with the disease remains an open question. Further research and prospective trials are warranted to address this issue.