







Pulmonary arterial hypertension (PAH) is a progressive disease of the pulmonary vasculature leading to right heart failure and death.1 Recent therapeutic advances include drugs targeting three separate signalling pathways2 involved in the pathogenesis of PAH. First the prostacyclin pathway evolves around prostaglandin I2 (prostacyclin),3 the main product of arachidonic acid in the vascular endothelium, which induces vascular relaxation by stimulating the production of cyclic adenosine monophosphate (cAMP) and inhibits smooth muscle cell growth.4 Several drugs were developed targeting the prostacyclin pathway: epoprostenol5 delivered by continuous intravenous infusion, inhaled or intravenous iloprost and either inhaled, subcutaneous or intravenous treprostinil. The second pathogenic pathway is the endothelin pathway. This involves endothelin-1, which has vasoconstrictor and proliferative effects on the smooth muscle cell. The effects of endothelin-1 are mediated through the endothelin A and endothelin B receptors. Bosentan6 and ambrisentan7 have been developed as endothelin receptor blocking agents. The third pathway targeted by PAH specific medications is the nitric oxide (NO) pathway. NO is a potent endogenous, endothelium derived, vasodilator that directly relaxes vascular smooth muscle through stimulation of soluble guanylate cyclase and increased production of intracellular cyclic guanosine monophosphate (cGMP).8 However, long-term inhaled nitric oxygen therapy is very cumbersome to use and an interruption of administration can cause haemodynamic deterioration.9 An alternative approved strategy of increasing activity of endogenous NO is to enhance nitric oxide-dependent cGMP-mediated pulmonary vasodilation through inhibition of phosphodiesterase type 5. Phosphodiesterase type 5 inhibitors such as oral sildenafil10 and tadalafil11 have been developed to achieve vasodilation in this manner. All drugs briefly discussed above were shown to improve exercise capacity and pulmonary haemodynamics with vasodilatory and antiproliferative effects on the pulmonary vasculature.2 Meta-analysis of randomised controlled trials shows that treatment with such targeted therapies also improves survival in PAH patients.12,13 Observational studies support this, with improved survival in patients treated in the modern management era compared with historical cohorts.14–16 However, despite the advent of these therapies PAH remains a progressive, fatal disease, with considerable room for improvement, especially for certain subgroups of patients, characterised by more severe outcomes. A number of factors have been associated with mortality in PAH. These include type of PAH (such as scleroderma associated and portopulmonary hypertension), presence of co-morbidities, functional status, exercise capacity at baseline, haemodynamic parameters (such as pulmonary vascular resistance [PVR]), mean right atrial pressure and cardiac output, but also male sex, renal function and brain natriuretic peptide levels.17 Of these parameters, functional status expressed as New York Heart Association (NYHA) functional class is one of the strongest predictors of outcome, not only at baseline, but also after medical treatments have been initiated.16 In addition to NYHA IV PAH, NYHA III patients with rapid progression of symptoms or severely compromised haemodynamics, such as right atrial pressure >15 mmHg or cardiac index <2.0 L·min-1·m-2, should be considered as severe disease.18 Currently, despite increasing PAH awareness in the community, a large number of patients are diagnosed with a more advanced disease.19 Untreated median survival in NYHA III PAH is 32 months and for NYHA IV patients this is further reduced to a mere six months.20 Given their poor prognosis and high rates of mortality optimal treatment should be initiated without further delay. For patients who persist in NYHA III and IV despite first-line monotherapy, survival is particularly poor and in these patients additional therapies are needed.16
The Paradox Between Haemodynamic Benefit and Clinical Deterioration
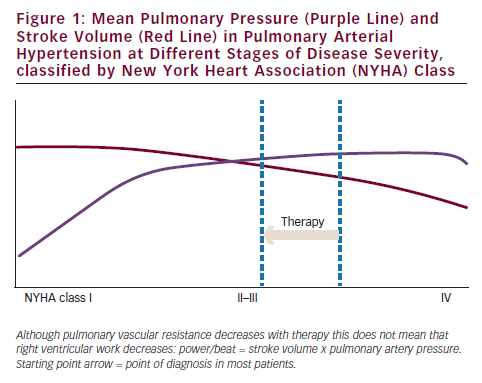
Right heart failure in pulmonary hypertension is primarily caused by increased afterload. In addition, right ventricular load is proportional to pulmonary vascular resistance if cardiac output is kept constant.21 It follows that normalisation of load will lead to restoration of the right ventricle as can be observed in patients with chronic thromboembolic pulmonary hypertension who received pulmonary endarterectomy.22 Based on this observation, one might expect that a modest reduction in PVR as observed in PAH patients who receive PAH-specific treatment will also automatically be followed by an improvement of right ventricular function.23 Based on the published clinical trials, it is fair to expect that a large proportion of patients do have an initial reduction in PVR and that this effect sustains over time.24 However, survival is still grim with a 55 % survival reported at three years in treated incident PAH. Also many patients continue to have marked limitations of normal daily activities.14 The question is: if we can achieve a reduction in PVR in the majority of patients, why is survival still worse? To study this further we investigated the relation between changes in right ventricular function and PVR in a large cohort of PAH patients. Results show a poor correlation between changes in right ventricular function, described as right ventricular ejection fraction, and PVR.25 In this study, changes in right ventricular ejection fraction were associated with survival, but changes in PVR were not, with similar changes in PVR in survivors versus non-survivors. So why can the right ventricle deteriorate even if PVR is lowered? Sniderman asked himself this question in the 1990s and suggested that the haemodynamic response of the right ventricle to a modest PVR reduction, which is an increase in cardiac output with almost no change in pulmonary artery pressure (see Figure 1), might explain this paradox. As a consequence of the unaltered pulmonary artery pressure, pathological wall tension on the right ventricle (defined as pressure times radius/two times wall thickness), will persist. Echocardiography and magnetic resonance imaging (MRI) studies in PAH studies showed that treatment has little impact on right ventricular diameter or volume during single agent treatment.24,26,27 In the case of a failing right ventricle at baseline, pathological wall tension will remain unaltered and the right ventricle might continue to fail. Indeed, haemodynamic data from the randomised controlled trials (RCTs) show that single agent has little impact on a reduction of pulmonary artery pressure. Figure 1 illustrates this further, describing the theoretical haemodynamic improvement that a patient will experience if diagnosed in advanced stage. The yardstick to improve right ventricular function according to Sniderman is thus to reduce pulmonary artery pressure.28 Another possible explanation is right ventricular power. Power per beat is defined by stroke volume times mean pulmonary artery pressure. In case a reduction in PVR leads to an increase in stroke volume without a decrease in pulmonary artery pressure, right ventricular power might even increase in case the load is reduced (see Figure 1). In fact, from the pulmonary artery pressure and stroke volume improvements reported in trials of PAH-specific medications it follows that these medications mostly increase power output of the right ventricle.23 For these two reasons, a modest reduction in right ventricular load in advanced disease will not automatically lead to improvement of right ventricular function and thus improved survival.
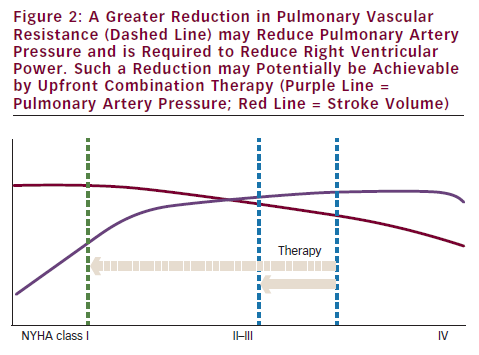
In early disease, the situation might be different. A reduction in PVR will in that case lead to a significant reduction in pulmonary artery pressure.29 However, since most patients are diagnosed in an advanced stage, the only solution to reduce right ventricular power and wall tension is to reduce PVR more (see Figure 2). The question remains whether this can be achieved by using current medications.
Can We Achieve a Reduction in Pulmonary Artery Pressure by Means of Current Medication?
Current guidelines recommend first-line monotherapy with agents targeting the prostacyclin, endothelin or NO pathway.1,2 Sequential combination therapy is currently recommended when clinical improvement fails to occur on monotherapy or further disease progression occurs.30 Epoprostenol is currently recommended as the treatment of choice for those patients with advanced disease.16 However, long-term survival in PAH treated with epoprostenol remains dismal, with 63 % survival at three years in a mixed cohort of incident and prevalent PAH patients.16 Though numerous clinical studies of combination therapy have hitherto been reported, most have examined the effect of sequential addition of treatment, and not upfront combination therapies. Only one trial studied upfront combination epoprostenol and bosentan versus epoprostenol and placebo – the Bosentan Randomized trial of Endothelin Antagonist Therapy for PAH (BREATHE-2) study. Improvements in PVR and mean pulmonary artery pressure (PAP) were greater in the upfront bosentan–epoprostenol treatment group, compared with the epoprostenol–placebo treated patients: respectively -35 % versus -26 % for PVR change and -9 % versus -2 % for mean PAP change. However, the difference did not reach statistical significance in this study, probably also due to the small sample size.31 In a larger but retrospective study, patients matched for age, NYHA functional class and PVR at diagnosis and treated with either first-line epoprostenol or first-line bosentan–epoprostenol were compared. Patient groups were compared for NYHA functional class and six-minute walk distance changes and pulmonary haemodynamic improvements assessed by right heart catheterisation at follow-up. In this study PVR improvement from treatment initiation to first follow-up was significantly greater in those who received the epoprostenol–bosentan combination therapy, compared with those receiving epoprostenol monotherapy: -48 % ± 17 % (± standard deviation [SD]) versus -29 % ± 17 %, respectively (p=0.0001). In this study a trend towards improved survival was described (p=0.07), emphasising the possible benefit of upfront combination therapy. Changes in exercise capacity and functional class were similar between combination and monotherapy patients in this study.32 From the same French study group a small series of 10 patients starting upfront triple combination therapy was reported – four NYHA class III and six NYHA class IV patients, with severely compromised haemodynamics (mean cardiac index 1.6 ± 0.4 L·L.min-1m2 and mean PVR 1,798 dyn.s-1cm5).31 For the seven patients with available data after four months of combination therapy, there was an impressive improvement in exercise capacity (from 290 ± 146 m at baseline to 454 ± 67 m during follow-up) and a 71 % reduction in PVR and normalisation of cardiac index. Mean PAP improved from 68 ± 17 mmHg at baseline to 45 ± 13 mmHg at four months (n=7; p<0.01) and 48 ± 10 mmHg at last visit (n=5; p<0.01). After a median 18.5 months follow-up (range 1–36 months) all patients were alive and in NYHA functional class I or II. In patients who were reassessed (n=5) at 12–28 months the positive findings established at four months were maintained.33 No further studies evaluating first-line combination therapy have been reported to date. Other studies have assessed the effect of combination therapy with prostanoids only in a sequential approach. The Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil (PACES) described the effects of the addition of oral sildenafil or placebo in patients stable on epoprostenol treatment.34 Those in the combination arm not only showed improvements in the six-minute walk distance and health-related quality of life scores, but also had longer times to clinical worsening and less need for epoprostenol dose increases. In this study haemodynamic changes were different with a -2.8 mmHg mean PAP decrease in combination therapy, compared with a +1.1 mmHg mean PAP increase with epoprostenol plus placebo (treatment difference -3.8 mmHg [95 % confidence interval (CI) -5.6 to -2.1]). PVR change was -151 dyn.s-1cm5 in combination therapy compared with +39 dyn.s-1cm5 in continued epoprostenol plus placebo. Relative increase in cardiac output was +0.9 L/min in patients with epoprostenol–sildenafil therapy (95 % CI 0.5 to 1.2 L/min). The iloprost inhalation solution Safety and pilot efficacy Trial in combination with bosentan for Evaluation in Pulmonary arterial hypertension (STEP) evaluated the addition of inhaled iloprost or placebo to ongoing bosentan therapy. In this trial patients randomised to iloprost demonstrated improvements in the six-minute walk distance, functional status, time to clinical worsening and decreased PVR.35 Further small non-randomised studies describe that oral sildenafil added to patients stable on bosentan leads to improved exercise capacity measured by six-minute walk distance and increased peak oxygen uptake (VO2 peak),36 sildenafil added to subcutaneous treprostinil improves treadmill times and dyspnoea fatigue scores,37 bosentan added to subcutaneous treprostinil leads to significant additional improvements in six-minute walk distance, Borg dyspnoea score and also decreases mean PAP further, from 56 ± 16 mmHg before bosentan start to 47 ± 12 mmHg (p<0.001), compared with 60 ± 15 mmHg at baseline, before treprostinil initiation.38 In another small descriptive study subcutaneous treprostinil or intravenous epoprostenol added to oral bosentan or oral bosentan–sildenafil improved six-minute walks, NYHA functional class and also improved right ventricular ejection fraction and decreased right ventricular end-diastolic volumes measured by MRI.39 Currently, it is unclear which of the possible combinations of PAH-specific therapies confer the greatest clinical gain. PAH-specific medical therapies are costly and cost-effectiveness of combining these therapies is uncertain.
Furthermore, the possibility of adverse events due to drug interactions is increased in the setting of combination therapy. However, the results of the studies are encouraging since they all show a larger decrease in PVR, and an indication exists that an even larger reduction in PVR can be achieved through upfront combination therapy. However, compelling evidence is lacking that upfront combination therapy is more advantageous compared with a sequential add-on strategy. Of interest in this respect is a large multicentre study comparing upfront ambrisentan in combination with tadalafil to ambrisentan monotherapy, using clinical worsening as an endpoint.40 Another study comparing upfront tadalafil and treprostinil versus treprostinil monotherapy was cancelled due to poor recruitment.41
Conclusion
Current treatments do lower PVR in the majority of patients and these effects are likely to sustain over time. However, this reduction in PVR might fail to halt the progression of right ventricular failure, since single agent therapy does not reduce right ventricular power, or right ventricular wall tension. Recent data from combination therapy offers the promise that a more pronounced effect on PVR and PAP can be reached. Whether this approach can also lead to a better improvement of right ventricular function and survival is still an open question.