










Major advances in heart failure (HF) management have been achieved over the past three decades, yet it remains a syndrome with high morbidity and mortality, poor quality of life and high healthcare costs. Despite all advances in medical/device therapy, many HF patients continue to deteriorate, leading to poor quality of life and high rehospitalisation and mortality rates.1 The recent European Society of Cardiology HF pilot study showed that hospitalised and stable/ambulatory HF patients had 12-month all-cause mortality rates of 17% and 7%, respectively, and 12-month rehospitalisation rates of 44% and 32%, respectively.2 Of note, an increase in left atrial (LA) pressure usually precedes the worsening of symptoms and acute HF decompensation in patients with left ventricular (LV) dysfunction.3 Acute HF has become the most common cause of hospitalisation in patients >65 years, and approximately 4–12% of patients present with cardiogenic shock, a severe condition with high morbidity and mortality rates.4,5
The creation of an interatrial shunt to decompress the left atrium in patients with acute or chronic HF has been used as an alternative therapy to improve symptoms and clinical outcomes.6–8 The aim of this review was to provide an updated overview and clinical perspective on interatrial shunting for treating HF, and to highlight the potential challenges and future directions of this therapy.
Interatrial Shunting for Acute Heart Failure
The management of cardiogenic shock includes cardiovascular resuscitation and identification and treatment of the underlying cause. Vasoactive agents are needed to maintain the cardiac output, but can increase myocardial oxygen demand leading to myocardial ischaemia and increased risk of arrhythmias. The use of mechanical circulatory support (MCS) can improve forward flow and interrupt the inflammatory mechanism preventing organ damage related to the shock. Veno-arterial extracorporeal membrane oxygenation (VA-ECMO) remains the most effective percutaneous MCS to treat refractory cardiogenic shock.9 Nevertheless, the arterial cannula may increase LV afterload in these patients, raising LV end-diastolic and LA pressures, leading to increased LV wall stress and myocardial oxygen consumption, and further worsening LV failure and refractory pulmonary oedema.10,11 The intra-aortic balloon pump has been the most common technique to decompress the LV in such cases, but it has not been associated with improved survival.12 Atrial septostomy (AS) has recently been described as an option for LV decompression in adults treated with VA-ECMO who develop refractory pulmonary oedema or upper body hypoxaemia.13
Clinical Evidence
The global experience of AS in patients treated with VA-ECMO is limited to small series and case reports. The main findings of AS in cardiogenic shock and VA-ECMO recipients to date are summarised in Table 1. AS was first described in paediatric patients treated with VA-ECMO who had signs of high LV/LA pressure or refractory pulmonary oedema. Haemodynamic data are limited, and some studies have evaluated the response of AS using non-invasive examintions, such as echocardiography, chest X-ray and respiratory parameters.
In a small series of four paediatric patients, the haemodynamic effect of AS was assessed by echocardiography and showed prompt normalisation of LA size and/or Doppler velocity values across the atrial septum, and improvements in LV function.14 A recent series of AS in 15 adults treated with VA-ECMO showed a significant drop in the oxygenation index (from 9.9 ± 5.9 to 4.6 ± 3.0, p<0.001) and radiological improvement of pulmonary oedema following AS (Figure 1). Additional LA pressure measurements obtained in three patients showed a significant decrease in LA pressure values following the procedure (from a mean of 29 mmHg to 13 mmHg).15,16 Another recent series of nine adult patients evaluated the radiological response after AS and a more objective measure using vascular pedicle width (VPW).17 VPW consists of the distance (measured in millimetres) from a perpendicular line at the point at which the left subclavian artery exits the aorta to the point at which the superior vena cava crosses the right main bronchus and correlates with volume overload.18 Significant radiological improvement and a reduction in VPW (from 76.6 mm to 57.9 mm, p=0.021) were observed following AS. Additionally, all patients had improved respiratory parameters, with an increase of PaO2/FiO2 of >10-fold, and a reduction in LA pressure (from 32 mmHg [interquartile range 28–42 mmHg] to 21 mmHg [interquartile range 13–28 mmHg]), and no changes in right atrial pressure.17
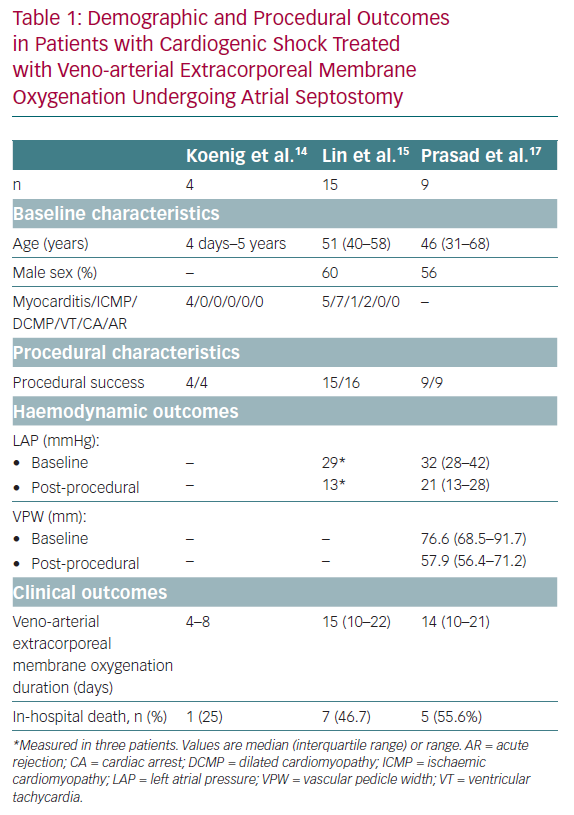
Overall, AS has been shown to be a relatively simple procedure with a high success rate among paediatric and adult patients treated with VA-ECMO who have refractory pulmonary oedema or upper body hypoxaemia.14–17 Apart from the acute effects of AS and the critical condition of patients receiving this treatment, the impact on clinical outcomes is difficult to establish due to the very limited number of patients and a lack of follow-up. LA decompression usually leads to a resolution of the pulmonary oedema and a shorter period of sedation and invasive ventilation. LV decompression improves subendocardial perfusion, decreases myocardial oxygen consumption and can contribute to LV recovery and ECMO weaning. While the reported in-hospital mortality following AS in such patients is approximately 50%, a significant proportion of patients who survive undergo successful heart transplantation.15,17 In summary, AS in this particularly challenging population acts as a palliative treatment for complications induced by VA-ECMO or cardiogenic shock to gain time for LV recovery or heart transplantation.
Technique
AS usually starts with a standard right heart catheterisation, with pressure and cardiac output measurements. A transseptal puncture is performed at the level of the central segment of the fossa ovalis under fluoroscopy and transoesophageal/intracardiac echocardiography guidance, followed by the advancement of a long sheath across the interatrial septum to the left atrium. A balloon is then advanced through a stiff guidewire, and balloon dilation at the level of the interatrial septum using a non-compliant balloon is performed. Usually, large balloons (>10–15 mm, up to 27 mm; usually an Inoue-Balloon or peripheral balloon) have been used in VA-ECMO patients to create a very large and haemodynamically effective atrial septal defect.
Future Perspectives
Patients with cardiogenic shock treated with VA-ECMO frequently need additional measures to decompress the LA. There are no data available comparing the different strategies to decompress the left heart, but surgical methods using a cannula connected to ECMO or the implant of another device, such as Impella, are likely to be more complex and expensive than AS. Also, it has been shown that bedside balloon AS in children undergoing echocardiographic guidance is feasible and prevents the transfer of these critical patients to the cath lab.14,19 In adult patients, AS has also been demonstrated to be simple, and could become one of the preferred strategies to overcome left heart distension in such patients. However, to the best of our knowledge, no prospective observational or randomised studies are underway to prospectively evaluate the efficacy of the AS strategy in the context of cardiogenic shock or VA-ECMO.
Interatrial Shunt for Chronic Heart Failure
HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF) continue to be major public healthcare problems with an annual cost of >US$30 billion in the US.20 The National Health and Nutritional Examination Survey estimates that 6.5 million Americans >20 years have HF, which is responsible for 900,000 hospitalisations every year.21 Furthermore, the projection from 2012 to 2030 estimates an increase close to 50%, resulting in more than 8 million people >18 years with HF.20 Despite decades of major advances in medical and device treatments, HF morbidity and mortality remain high, regardless of their aetiology.
Increased LA pressure leading to pulmonary congestion is the common mechanism precipitating worsening of symptoms and acute decompensation in chronic HF patients.3 In addition, elevated pulmonary capillary wedge pressure (PCWP) during exercise has been associated with lower functional capacity, negatively impacting quality of life and prognosis.22,23 Implantable haemodynamic pressure monitoring has been demonstrated to decrease HF hospitalisation and improve outcomes by guiding dose titration of drugs, impacting LA pressure in HFrEF and HFpEF patients.3,24 Notwithstanding the benefits of device-guided therapy, most HF patients are elderly and present multiple comorbidities, which may challenge both the self-titration of medication and close medical follow-up.
There is some evidence supporting interatrial shunting to relieve the volume excess from the left to right atrium regulated by the interatrial pressure gradient, and creating an on-demand, auto-regulating reduction in LA pressure. Pulmonary oedema due to decreased LV compliance and acute increase in LA pressure has been observed in patients undergoing atrial septal defect closure.25 One of the concerns of left-to-right shunting is the potential overloading of the right ventricle (RV) and an increase in pulmonary artery pressure (PAP). However, studies in the congenital field have shown that small shunts (usually atrial septal defects <10 mm) are not associated with any deleterious haemodynamic effect at long-term follow-up.26 Thus, some devices have been developed in order to create a controlled and permanent left-to-right shunt in HF patients.
Devices and Clinical Evidence
InterAtrial Shunt Device
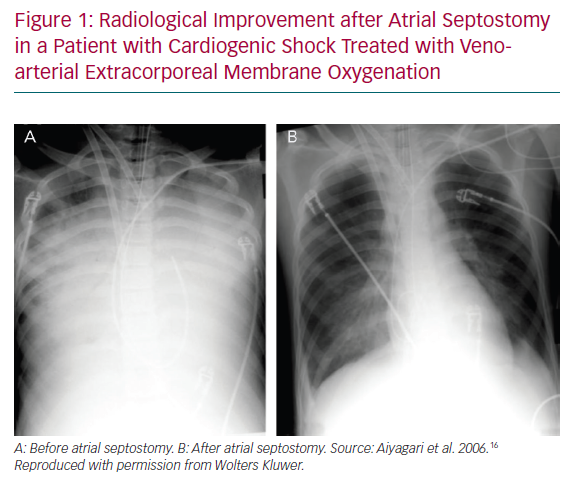
The InterAtrial Shunt Device (IASD, Corvia Medical) is composed of a nitinol mesh with multiple legs and radiopaque markers, with a central hole (Figure 2A and Figure 2B). The disc and fenestration diameter are 19 mm and 8 mm, respectively.27
The IASD has been evaluated in patients with HFpEF. In the first evaluation of 11 patients with left ventricular ejection fraction (LVEF) >45% and New York Heart Association (NYHA) class III/IV, the device was successfully implanted in all patients (in one case, the device was initially maldeployed in the left atrium, recaptured with a snare without complications and a second device was successfully implanted).27 Following device implantation, there was a significant decrease (about 30%) in PCWP, with stable right atrial pressure (RAP) and PAP. This was associated with significant improvements in a 6-minute walk test (6MWT) distance, quality of life and NYHA class at 30-day follow-up (Table 2).27
The Reduce Elevated Left Atrial Pressure in Patients with HF (REDUCE LAP-HF) trial was a multicentre study of 64 patients with symptomatic HF (NYHA classes II–IV) and LVEF >40% who were treated with the IASD System II device.28 The device was successfully implanted in all patients, with no major periprocedural complications. At 6-month follow-up, repeat right catheterisation showed no significant changes in PCWP at rest, but there was a significant decrease in PCWP at peak exercise (mean reduction of 3 mmHg, p=0.01). There were no safety issues, and significant improvements in NYHA class, quality of life and exercise capacity were observed at 6 months (Table 2).28 A subsequent report from Kaye et al. showed that these improvements were maintained at 1-year follow-up.29 Shunt patency was properly evaluated by transthoracic echocardiography (TTE) in 48 of 64 patients, with no signs of occlusion/stenosis. In 16 patients (25%), TTE images were not considered adequate for patency assessment, highlighting the potential challenge of interatrial shunt evaluation using TTE. Recently, Kaye et al. determined the potential impact of IASD implantation on mortality in HFpEF patients after a median follow-up of 2 years.30 The observed mortality in IASD recipients (9.4%) was compared with the predicted mortality using the Meta-analysis Global Group in Chronic HF prognostic model at 1 and 3 years, showing that interatrial shunting was associated with a 33% reduction in the all-cause mortality rate (HR 0.67, 95% CI [0.09–0.89], p=0.02). Furthermore, IASD recipients who had HF hospitalisations were not associated with subsequent increased mortality (p=0.31).

The REDUCE LAP-HF I study was a randomised trial that included patients with LVEF >40% and NYHA classes III–IV.8 Forty-four patients were allocated (1:1) to receive the IASD System II device versus a sham procedure (control group). At 1-month follow-up, there were no significant differences between the groups for PCWP values at rest, but patients in the IASD group had a reduction of PCWP during exercise compared to a lack of changes in PCWP exercise values among the control group (p=0.01 for differences between groups; Table 2). Functional status and exercise capacity were similar in both groups at 1-month and 1-year follow-up.8,31 At 1-year follow-up, the IASD group exhibited a tendency towards a better NYHA class improvement compared to baseline (median −1 [interquartile range −1 to 0] versus 0 [−1 to 0], p=0.08) and less HF hospitalisations (rate per patient year 0.22, 95% CI [0.08–0.58] versus 0.63, 95% CI [0.33–1.21], p=0.06).31 Mild RV dilation was observed at 6 months in the IASD group compared with the control group (9.1 ml/m2 [interquartile range 5.8–11.0] versus −1.9 ml/m2 [interquartile range −4.4 to 3.8] , p=0.002), with no further dilation up to 1 year. No decrease in RV function and no evidence of shunt stenosis/occlusion were observed over time. Finally, major adverse cardiac, cerebrovascular or renal events were also similar between the groups, with a survival rate of 95% at 12 months (one death in each group).
Additional evidence of the potential effect of interatrial shunting on right-side heart parameters was recently reported by Danial et al.32 An interatrial shunt was created with a 2 mm balloon in a total of 22 rats (healthy n=11, HFpEF n=11), and this translated into a reduction in LA volume and pulmonary artery diameter in rats with HFpEF. However, in healthy rats, interatrial shunting was associated with an RV overload and increased pulmonary artery diameter. Long-term clinical data are needed to provide definitive data on the potential impact of interatrial shunts on right ventricular function.
V-Wave Device
The V-Wave device is an hourglass-shaped device made with nitinol and expanded polytetrafluoroethylene encapsulation, and three porcine pericardial leaflets sutured together with a PROLENE suture to ensure unidirectional left-to-right shunt (Figure 2C and Figure 2D). The lumen diameter of the V-Wave device is 5 mm.7 This device has been tested in patients with HFpEF and HFrEF. The newer generation device consists of a similar device, but without valve leaflets (valveless device) (Figure 2E and Figure 2F). After an initial in-human experience, this newer generation device will be used in a large randomised trial of HFrEF and HFpEF patients.
The V-Wave device was the first interatrial shunt device implanted in a patient with HFrEF.33 Del Trigo et al. reported the initial experience with this device in 10 patients with HFrEF (mean LVEF 25%) and functional class III/IV, despite optimal medical/device therapy.7 The device was implanted in all patients with no complications, and a significant reduction in PCWP (mean 5 mmHg) was observed at 3-month follow-up, with no changes in RAP or PAP values. At 3-month follow-up, most patients were in NYHA classes I–II, with significant improvements in quality of life, as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ; 44.8 ± 9.4 versus 79.1 ± 13.0, p<0.001), and the 6MWT distance increased by about 75 m (p=0.01 versus baseline).
The results of an initial multicentre experience with the V-Wave device were recently reported.34 The study included 38 patients (HFrEF n=30, HFpEF n=8) with NYHA class III–IV, despite optimal medical/device therapy.The V-Wave device was successfully implanted in all cases with only one major periprocedural complication (cardiac tamponade resolved with pericardiocentesis). No additional device-procedure related events occurred up to 1-year follow-up (primary endpoint). Significant improvements in functional status, exercise capacity and quality of life were observed within the first 3 months post-procedure, and were maintained at 1-year follow-up. There were no changes in haemodynamic parameters (as determined at rest) at 1-year follow-up, including PCWP, RAP and PAP (Table 2). After a median follow-up of 28 months (21–31 months), 10 patients (26%) had died (eight from cardiovascular causes), one patient received a left ventricular assist device as destination therapy at 15 months and another underwent heart transplantation at 27 months.
The patency of the valved V-Wave device was evaluated by transoesophageal echocardiography at two time points: at 1–3 months and at 1 year after device implantation.34 All shunts were fully patent at 1–3 months, but shunt occlusion was observed in 14% of patients at 1-year follow-up. Additionally, some degree of shunt stenosis at the valve level occurred in 36% of patients, leading to an incidence of shunt stenosis or occlusion of 50% at 1 year. No thrombus (as evaluated by echocardiography) was detected in any of the patients. Interestingly, the potential cause of stenosis or occlusion was suggested from a stenotic shunt that was explanted during cardiac transplantation about 2 years after the procedure. The bioprosthetic leaflets were thickened and stenotic with neoendocardial hyperplasia (pannus). These data, along with the lack of thrombus, strongly suggest intrashunt valve deterioration as the main mechanism of shunt stenosis–occlusion. Following this initial experience, modifications were implemented in order to improve late device patency and continued efficacy, with valve removal being the most relevant feature of the newer (second) generation of the V-Wave shunt. In an animal model of 11 sheep, the valveless V-Wave shunt remained patent, with no late loss in lumen diameter at 5–6-month follow-up (V-Wave, unpublished data). In addition, the first-in-human experience with the valveless V-Wave shunt showed the patency of the shunt (no stenosis-occlusion) in all cases at the 1-year follow-up.35
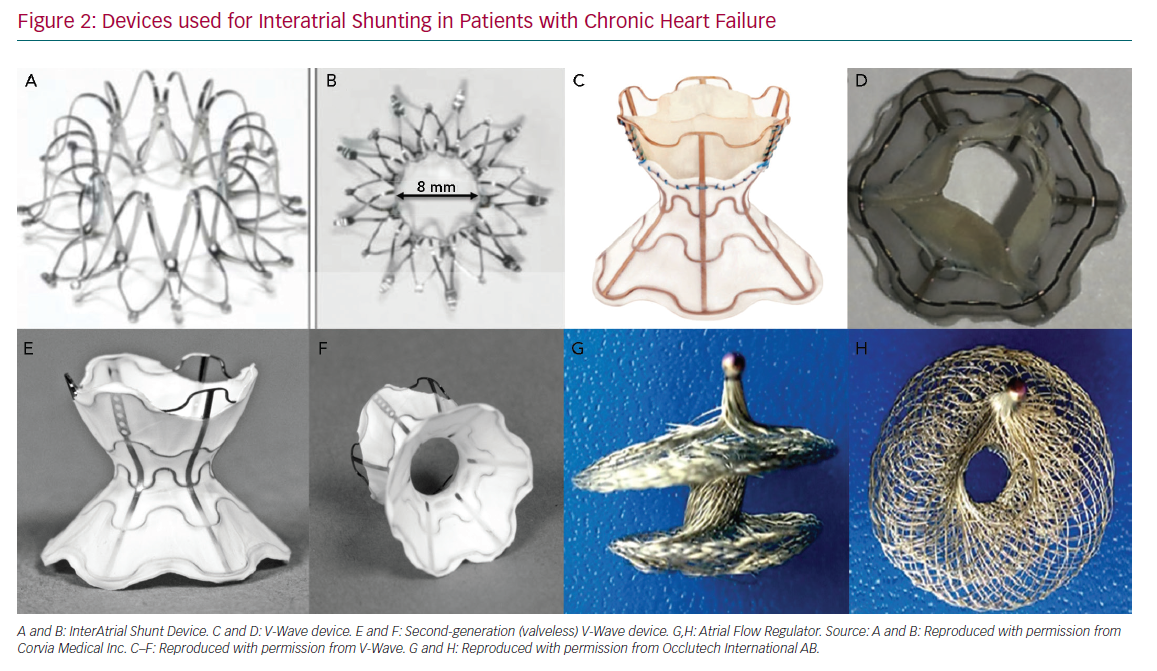
The fact that half of the interatrial shunts were either stenotic or occluded in the initial V-Wave experience permitted a comparative analysis between patients with and without shunt stenosis or occlusion.34 Patients with full patent shunts exhibited significant improvements in haemodynamic parameters, such as PWCP, at follow-up (mean decrease of 4 mmHg, p=0.01 versus baseline), compared to the lack of changes in the shunt stenosis–occlusion group (p=0.03 for the comparison of PWCP changes between groups). Furthermore, patients with patent shunts had improved late clinical outcomes, with lower rates of death/left ventricular assistance/transplantation or HF rehospitalisation at 3-year follow-up (Figure 3). Of note, the Kaplan–Meier curves suggested relatively similar event rates (death, hospitalisation) for the two groups (patent versus stenotic-occluded shunts) during the first year of follow-up, before curves progressively separated at up to 3 years. In an exploratory analysis, clinical outcomes at up to 3-year follow-up in those patients included in the V-Wave initial experience appeared to be better than those observed in the CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Heart Failure Patients (CHAMPION) trial, which had similar inclusion criteria and patient characteristics, particularly among those patients with full patent shunt at 1-year follow-up, even if the V-Wave group patients were older and exhibited a higher risk profile (Supplementary Material Figure 1).36
Atrial Flow Regulator
The Atrial Flow Regulator (AFR; Occlutech) is a double disc device made of a nitinol wire mesh and a central orifice (Figure 2G and Figure 2H). The fenestration diameter varies from 4 to 10 mm, and there are three waist sizes: 2, 5 and 10 mm to suit the atrial septal thickness. The AFR device is being tested in patients with HFrEF and HFpEF in a small feasibility, currently ongoing clinical trial (Pilot Study to Assess Safety and Efficacy of a Novel Atrial Flow Regulator in Heart Failure Patients [PRELIEVE], NCT03030274).
In summary, all of the tested devices to date have been demonstrated to be safe, with improvements in functional class and quality of life, despite a modest reduction in absolute wedge pressure values compared to baseline.
Technique
For most devices, the procedure is performed under general or local anaesthesia, by transfemoral venous approach. Following transseptal puncture, a sheath (14–16 Fr) is advanced into the LA cavity, and the device is deployed using a dedicated delivery system. The left side of the device is initially opened, and the entire system is pulled back ensuring back tenting at the level of the interatrial septum. Then, the right side of the device is deployed and the device is finally released.
Following device implantation, most studies to date have recommended aspirin associated with clopidogrel for 6 months in patients who are not under anticoagulation therapy, and in patients receiving anticoagulantion (warfarin or direct oral anticoagulant), aspirin is added to the antithrombotic regime.
Future Studies
The ongoing and future studies on interatrial shunting in patients with HF are summarised in Supplementary Material Table 1. Several studies on the IASD are currently in the recruitment phase. The REDUCE LAP-HFrEF trial (NCT03093961) is recruiting up to 10 HFrEF patients in NYHA classes III–IV to evaluate the safety and feasibility of the device in this specific population.
Another study involving the IASD, the REDUCE LAP-HF II (NCT03088033) is a randomised controlled trial comparing the clinical efficacy of the device versus optimal medical treatment (with a sham procedure) in symptomatic patients with LVEF >40%. The estimated sample size consisted of 608 patients, with the primary composite endpoint of the incidence of and time-to-cardiovascular mortality or first non-fatal ischaemic stroke within 12 months; total rate (first plus recurrent) per patient year of HF admissions or healthcare facility visits for IV diuresis for HF within 12 months and time to first HF event; and change in baseline KCCQ total summary score at 12 months. Finally, the REDUCE LAP-HF III (NCT03191656) is planning to recruit 100 patients with preserved or mildly reduced LVEF, with the purpose of determining the benefits (functional status, quality of life) of this therapy at 12-month follow-up.
Reducing Lung Congestion Symptoms in Advanced Heart Failure (RELIEVE-HF, NCT03499236) is a randomised controlled trial comparing the second-generation (valveless) V-Wave device with optimal medical therapy (with a sham procedure) in 500 patients with HFpEF or HFrEF. The primary endpoint is a hierarchical composite of death, heart transplant or left ventricular assist device implantation, HF hospitalisations and change in 6MWT (time frame: follow-up duration at endpoint analysis ranges from a minimum of 12 to a maximum of 24 months).
Conclusion
Interatrial shunting has emerged as a promising option for treating HF patients. The feasibility and acute efficacy of balloon dilation atrial septostomy have been shown in acute HF, particularly among patients with refractory pulmonary oedema treated with VA-ECMO. To date, clinical experience with interatrial shunting in chronic HF (HFrEF and HFpEF) has been limited to less than 200 patients using different devices, and demonstrating to be a feasible and safe therapy in those who remain symptomatic, despite optimal medical/device therapy. If further randomised trials (currently ongoing) could demonstrate improvements in clinical outcomes, device-mediated left-to-right atrial shunting would become an important new approach for treating patients with refractory HF.
Click here to view Supplementary material.