Cardiac resynchronisation therapy (CRT) has been a cornerstone in the treatment of select advanced heart failure cases since its introduction to our armamentarium in the early 2000s.1 Indeed, 30–60% of advanced heart failure patients exhibit evidence of dyssynchrony, when defined electrocardiographically or mechanically.2–4 The latter is a consequence of the former. CRT has several unique features, summarised in Table 1.
Although the main mechanisms through which CRT is thought to act are improvements in chamber mechanics, a more subtle effect has been recognised, linking resynchronisation to cellular metabolism and energy efficiency.
Full comprehension of this connection could help us to interpret non-response and lead to more sophisticated criteria for CRT use, help detect concealed responders – patients who do not have increases in mechanical output but do have improvements in bioenergetics and may have improved functional reserve – and alter CRT programming approach.
This article will present cellular bioenergetics in myocardial cells and subsequent alterations in heart failure. Cellular and molecular aspects of CRT effects on bioenergetics will be discussed, along with their potential implications for clinical practice.
Cardiomyocyte Metabolism in Health and Heart Failure
Normal Conditions
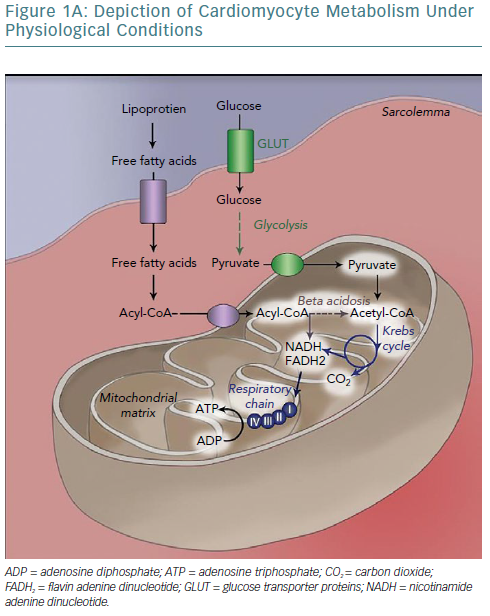
The human heart has been called a metabolic omnivore because of its ability to use all common oxidative substrates. Under normal conditions, 60–90% of adenosine triphosphate (ATP) produced is a product of fatty acid oxidation with the rest attributed to glucose oxidation.5 Notably, these metabolic pathways are mutually inhibitory (the Randle cycle) and the relative flow is determined by the fed or fasted state of the cell; fed leading to preferential use of glucose and fasted to fatty acid oxidation.6 Given the much higher efficiency of aerobic oxidation compared with anaerobic metabolism, the heart predictably uses the former (it accounts for 90% of energy production).7,8 Oxidative metabolism begins in the sarcoplasm but continues in the mitochondria, which occupy 30% of cardiomyocyte volume, strategically placed near myofibrils and sarcoplasmic reticulum (SR) to minimise the diffusion distance of the high-energy bond-containing molecules from production to consumption sites.8 This highlights the crucial dependency of cellular function on mitochondrial integrity, placement and output.
The burden of cardiac work is such that there is no energy production reserve. At maximal exercise the heart is operating at >90% of maximum oxidative capacity and recycles an amount of ATP (6 kg) more than 20 times its weight.6,9


As such, establishing effective coupling between energy producing and consuming organelles is crucial. Calcium ions are a strong candidate for this role inasmuch as they participate in both actin-myosin crosstalk regulation and in activation of major mitochondrial complexes (Table 2) and ATP synthase.10–12
One of the key aspects of cellular bioenergetics lies in the recognition that the sarcoplasm is not a homogeneous solution of molecules; rather, local concentrations of metabolites are essential in determining metabolic efficiency, as dictated by the laws of thermodynamics.7 More specifically, the energy yield from ATP dissociation is determined by the equation:

Where ΔG denotes change in Gibbs free energy, a measure of the energy yield and so of the spontaneous nature of a reaction, combining both enthalpy (thermal output) and entropy (measure of randomness) through the equation:

With H denoting enthalpy, T absolute temperature and S entropy. ΔG0 is the standard free energy change at a temperature of 25°C (298 K), R is the universal gas constant in J mol-1 K-1, ln(X) is the natural logarithm of X and [Y] denotes the local concentration – allowed to freely diffuse in the vicinity without barriers such as membranes – of substance Y.
Consequently, the free energy yield (ΔG) is critically dependent on local concentrations of products and reactants and fast regeneration of ATP, along with rapid removal of adenosine diphosphate (ADP) from the vicinity to ensure optimal energy output. Simply put, the same intracellular task may require more ATP as a result of lower energy from every ATP dissociation owing to the mismanagement of reactants. Reduced yield may cause reactivation of the foetal myosin isoform (beta-myosin), producing less shortening per power stroke, but requiring less energy to undergo conformational changes than the adult one (alpha-myosin).13
In addition to the electron transport chain/ATP synthase proximity in mitochondria, all the enzymes of glycolysis are organised in complexes attached to energy-consuming structures (SR/myofibrils). Phosphocreatine (PCr) kinase also exhibits high concentrations near myosin head regions, SR calcium transport ATPase (SERCA) pumps and ADP/ATP mitochondrial antiporter, where it ensures rapid ATP regeneration and – in the latter case – rapid ATP shuttling to the sarcoplasm, maintaining optimal yield from ATP dissociation.14–17
Prerequisites for optimal and efficient cellular bioenergetics are listed in Table 3.
Alterations in Heart Failure
In heart failure, the heart switches from an omnivore to preferentially using glucose oxidation for energy production, initially by increasing glucose consumption and ultimately by decreasing fatty acid oxidation.18–20 In the short term, glucose and fatty acids compete with each other for use as substrates for energy production (the Randle cycle).21 In the fasted state, beta-oxidation of the relatively abundant fatty acids – released by the liver – causes an increase in the mitochondrial ratios of acetyl-coenzyme A/coenzyme A and reduced nicotinamide adenine dinucleotide (NADH)/oxidised nicotinamide adenine dinucleotide (NAD+), which both inhibit pyruvate dehydrogenase activity.22 Conversely, in the fed state, malonyl-coenzyme A, a by-product of glucose oxidation, inhibits fatty acid oxidation and promotes lipogenesis by inhibiting carnitine palmitoyltransferase.23 It appears that in heart failure, stressors – in the form of oxidative stress or adrenergic signalling – continuously promote glucose use as a substrate for energy production, because of its effects beyond ATP production.24
In the long term, changes in enzyme levels or activity are involved in regulating substrate use. Indeed, enzymes involved in fatty acid oxidation are downregulated in heart failure; a return to the foetal pattern.25,26 More specifically, entry of glucose catabolism products into the Krebs cycle is facilitated through a reduction of pyruvate dehydrogenase kinase – which phosphorylates and inhibits pyruvate dehydrogenase, necessary for the conversion of pyruvate to acetyl-coenzyme A – while transcript levels of carnitine palmitoyltransferase – necessary for acylcarnitine reconversion into acyl-coenzyme A and carnitine after entry into mitochondria – are downregulated.25,27 Furthermore, beta-oxidation is itself inhibited by reduced levels of some acyl-coenzyme A dehydrogenases, both in foetal and failing hearts.
Citrate synthase messenger RNA levels are also found to be reduced in the foetal gene-expression pattern, leading not merely to a metabolism based on glucose, but more specifically a glycolytic one. Increased glucose availability – at least prior to development of insulin resistance – may underlie the myosin isoform switch, through O-glycosylation of transcriptional factors.28 Furthermore, the foetal pattern promotes cell survival by means of innate antiapoptotic pathway activation, such as that mediated by the protein kinase B/mammalian target of rapamycin.29 The exact triggers for this switch are not well understood, but it is thought that exposure to a hypoxic milieu – caused, in heart failure, by vascular disease, fibrosis and increased workload – is reminiscent of the in utero environment and is the underlying cause of this change, leading to faster adaptation to changing stimuli mediated by a multitude of mechanisms acting on both the transcriptional and epigenetic level.28,30
The main activator of fatty acid oxidation is peroxisome proliferator-activated receptor-alpha (PPAR-alpha), which increases expression of genes involved in the entry, transport to mitochondria and beta-oxidation of fatty acids and downregulates expression of genes of proteins participating in glucose metabolism. Reduced PPAR-alpha expression has been described in heart failure and has been linked to fibrosis and mitochondrial fragmentation.31 This phenomenon may represent an adaptational attempt given that, stoichiometrically, the amount of oxygen consumed per ATP produced is higher for fatty acids than for glucose.32 Potential causes of lipotoxicity are detailed in Table 4.
In fact, increased glycolytic capacity has been related to increased survival in heart failure.33 Furthermore, localisation of glycolytic enzymes in the sarcoplasm and a preferentially glycolytic metabolism allows for ATP use in housekeeping processes – such as sodium–potassium ATPases and SERCAs – and provides substrates for the pentose phosphate pathway. The pentose phosphate pathway is critical for synthesis of antioxidants such as NADPH, allowing cellular survival – maintaining proper ionic concentrations and membrane potential – albeit at the cost of functionality.24,34 This is advantageous in the short term but detrimental in chronic conditions. Given the much higher amount of ATP produced per fatty acid molecule, many glucose molecules must be oxidated to match this yield. However, in advanced heart failure a reduced glucose oxidation capacity is found, primarily attributed to the insulin resistance of the failing heart because of an increased concentration of non-metabolised fatty acids and sympathetic system and renin-angiotensin-aldosterone axis activation.25,35–37 Downregulation of sarcoplasmic membrane glucose transporters has also been noted.38 This is especially true for septal segments in dyssynchronous heart failure where decreased workload – contraction against relaxed lateral segments – allows for reduced glucose consumption.39,40 Moreover, beta-oxidation leads to the production of Krebs cycle intermediates (anaplerotic reactions).33 Obviously this deprives the heart of its fuel, leading to energy starvation.9


As such, myocardial substrate metabolism tuning crucially affects left ventricular energetics in vivo, to the extent that preservation of fatty acid metabolism preference has been found to be linked with lack of clinical response to CRT.41,42 A twofold explanation may be given; either the failing myocardium has not switched to glucose metabolism as a compensatory mechanism and so continues to use an ineffective fuel, or full metabolic compensation at the microscopic level has been achieved so normal preference to fatty acids is maintained and no potential for further improvement exists.43 The latter interpretation is more likely given that CRT has been found to increase fatty acid metabolism, in itself facilitating oxidative metabolism.
PCr kinase levels are decreased in heart failure.44,45 This leads to a lower ATP/ADP ratio which – aside from the obvious negative effects discussed previously – may serve an unexpected adaptive purpose. That is, ensuring – by the opening of ATP-gated potassium channels of the mitochondrial membrane, which hyperpolarise the organelle – that cytochrome c will not be released and apoptotic processes will not be triggered.46 Furthermore, the mitochondrial isoform of PCr kinase partakes in a feedback mechanism ensuring that energy production is matched to consumption; ADP from the sarcoplasm is pumped into the mitochondria by the adenosine nucleotide transporter, converted to ATP by the mitochondrial PCr kinase and shuttled back through the same path. Loss of PCr means that the mitochondria can no longer respond accordingly to increased sarcoplasmic ADP.47
A complementary ATP-replenishing system involves adenylate kinase, which converts two ADP molecules into ATP and adenosine monophosphate (AMP). In addition, increased AMP levels lead to AMP-dependent protein kinase (AMPK) activation, which constitutes a central element of adaptational mechanisms in low-fuel conditions.48 Specifically, AMPK, when acutely activated, shuts down ATP-consuming processes such as fatty acid and glucose synthesis, activates ATP-producing pathways such as fatty acid oxidation and glucose intake through glucose transporter type 4, and increases insulin sensitivity, ensuring cellular survival.17,48–50 However, chronic activation is detrimental because fatty acid metabolism is reduced through mitochondrial transport inhibition and apoptosis is activated.51
Heart failure is a state of severe energy wasting, i.e. energy consumption not leading to useful work but lost as heat.52 Increased uncoupling protein concentration has been observed, leading to proton leak back into the matrix.9,33 Altered calcium homeostasis may lead to the myosin head performing the power stroke while unbound to actin (troponin C dysfunction), so not leading to sarcomere shortening.53,54 Finally, abnormalities in proteins connecting sarcomeres to the extracellular matrix lead to decoupling and prevent sarcomere shortening translating into cardiomyocyte contraction.55
Mitochondrial dysfunction in heart failure merits further consideration. Increased reactive oxygen species (ROS) concentrations in heart failure lead to electron transport chain dysfunction and damage mitochondrial proteins and genome.34,46 However, a burst of ROS may actually prove beneficial inasmuch as it triggers activation of the master regulator of mitochondrial biogenesis and oxidative capacity; peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1). This protein allows for increased expression of nucleus-encoded mitochondrial transcription factor 1, increasing mitochondrial biogenesis, oxidative capacity and fatty acid use.8,56 Furthermore, PGC-1 increases synthesis of antioxidant enzymes (catalase, superoxide dismutase and glutathione peroxidase) in response to redox signalling.57
Chronically elevated catecholamine and angiotensin levels and increased TNF-alpha and endothelin-1 lead to protein kinase B activation, which, in turn, downregulates PGC-1, causing mitochondrial fragmentation (inability to replicate) and decreased concentrations of electron transport chain complexes and ATP synthase.5,7,9,10,33 Although CRT does increase protein kinase B phosphorylation/activity, this should be viewed in light of global prosurvival effects of this kinase, rather than its adverse actions on cellular bioenergetics, which are mitigated through different pathways by resynchronisation.58,59 Finally, and especially pertaining to dyssynchronous heart failure, with abnormal stretching of non-activated cells, an intimate crosstalk exists between cardiomyocyte stretch and apoptosis. This appears to be mediated by mitochondria, as evidenced by proapoptotic molecule (p53 and Bax) increases, leading to mitochondrial membrane depolarisation and release of cytochrome c and other complexes triggering apoptosis upon cellular stretching.60
In short, teleologically, heart failure initially redirects cardiomyocyte metabolism towards glucose use, which ensures cellular survival at the cost of function. However, insulin resistance in advanced stages of heart failure prevents use of glucose as efficiently.61 Studies have suggested that the failing heart may attempt to switch to ketone bodies oxidation in an effort to sustain its metabolism and increase its efficiency (conversion into usable substrate not requiring much energy).27,62,63 Ketone bodies oxidation is a rapidly usable and relatively dense – requiring two steps to enter the Krebs cycle as acetyl-coenzyme A and each yielding two acetyl-coenzyme A molecules – source of energy, supplemented by the liver.
Branched chain amino acid catabolic pathways have been found to be downregulated in a murine heart failure model – with reduced expression of key enzymes such as the branched-chain alpha-keto acid dehydrogenase complex – presumably because of oversupply of ketone bodies by the liver (two out of three proteinogenic branched chain amino acids are ketogenic).64 However, this leads to an accumulation of branched-chain keto acids, which induces inhibition of mitochondrial respiration.65 Moreover, factors promoting fatty acid oxidation (PPAR-alpha and PGC-1) are also regulators of cellular oxidative capacity in general.34 As such, their downregulation leads not only to preferential glucose use but also to reduced oxidative potential of the cell. Several of the previously mentioned disturbances are (partially) reversed by effective resynchronisation, as discussed in the following sections.
Chamber Mechanics and Energy Consumption in Dyssynchronous Heart Failure
It should be noted that most studies attempting to clarify effects of resynchronisation on chamber mechanics, substrate metabolism and energy efficiency have been conducted in dilated cardiomyopathy models with concomitant left bundle branch block because of the perceived homogeneous myocardial involvement that leads to more predictable behaviour, i.e. excitation sequence, after CRT initiation. Conversely, ischaemic cardiomyopathy of such severity (ejection fraction ≤35%) usually involves the presence of extensive dense scar regions, which critically modify electrophysiological properties (the stimulus may need to bypass a fixed line of block and, in the process, depolarise different myocardial segments). This is reflected by ischaemic aetiology being considered a negative prognosticator for response to CRT.66–74 The patients expected to gain the greatest benefits from chamber resynchronisation are those with the most pronounced basal dyssynchrony, exhibiting the widest QRS complexes.75–77
Modern views on dyssynchronous heart failure include the notion of adverse mechanoenergetics, leading to reduced energy efficiency, evidenced by a reduced ratio between energy consumed (in the form of oxygen) and external work performed (stroke work).78–80 Several explanations can be offered (Table 5).
Compared with inotropes, CRT has been found to acutely improve systolic function (increased ) while reducing oxygen consumption assessed by arterial/coronary sinus oxygen difference and minute oxygen consumption).81 Chamber segments resynchronisation offers a plausible interpretation – by negating increased interior work – for improved energetics upon CRT introduction. An additional beneficial effect of CRT could lie in the reversal of the shortening of diastolic period and diastolic dysfunction, both present in dyssynchronous heart failure in general and particularly so in the left bundle branch block variety.82–84 More specifically, divergence in the timing of systole between myocardial segments in left bundle branch block dilated cardiomyopathy has been shown to worsen microvascular function of the left anterior descending artery territory (perfusion occurring during diastole) and consequently flow reserve, raising the potential for therapeutic interventions.85 In the mechanoenergetics context, although not improving efficiency, CRT could improve overall available energy by increasing oxygen and substrate delivery to myocardium.
) while reducing oxygen consumption assessed by arterial/coronary sinus oxygen difference and minute oxygen consumption).81 Chamber segments resynchronisation offers a plausible interpretation – by negating increased interior work – for improved energetics upon CRT introduction. An additional beneficial effect of CRT could lie in the reversal of the shortening of diastolic period and diastolic dysfunction, both present in dyssynchronous heart failure in general and particularly so in the left bundle branch block variety.82–84 More specifically, divergence in the timing of systole between myocardial segments in left bundle branch block dilated cardiomyopathy has been shown to worsen microvascular function of the left anterior descending artery territory (perfusion occurring during diastole) and consequently flow reserve, raising the potential for therapeutic interventions.85 In the mechanoenergetics context, although not improving efficiency, CRT could improve overall available energy by increasing oxygen and substrate delivery to myocardium.
The most robust method of assessing substrate metabolism relies on the use of glucose and fatty acid radioactive isotopes and their in vivo turnover rates.43,86–89 Substrate metabolism, but not perfusion, is also altered in dyssynchronous heart failure. The septal wall uses less glucose than lateral segments, potentially owing to the preferential conversion of mechanical work to stretching of inactive myocardium, as opposed to blood expulsion through the aortic valve, mediated molecularly by reduced glucose transporter expression.38,90,91 Conversely, upon contraction, the lateral wall consumes higher amounts of glucose-derived energy because of its pre-stretched condition (exceeding Frank-Starling law limits). Furthermore, this is in accordance with preferential glucose use in advanced heart failure, as the lateral wall could be considered to be at a more locally advanced heart failure stage.
Consequently, glucose uptake by 13-fluorodeoxyglucose – measured by PET – may represent increases in glycolysis rather than oxidative phosphorylation and prioritisation of survival over function. As such, this pendulum-like internal energy transfer accounts for significant macroscopic causes of reduced mechanical efficiency. When oxidative metabolism is specifically assessed by means of acetate clearance – a precursor to acetyl-coenzyme A that enters the Krebs cycle – an increase in septal and a decrease in lateral oxidative metabolism upon CRT was noted, leading to global homogenisation without increases in metabolic demand, despite increases in mechanical work output.92 This appears to contradict previous interpretations of CRT effects on substrate metabolism, yet it can be hypothesised that the increased septal work reactivated oxidative phosphorylation to increase energy output, whereas the lateral wall operates at less-than-maximal oxidative capacity and can reduce oxidative metabolism, also reducing ROS burden in the process.
Predictably, CRT has been found to rectify and homogenise glucose use throughout the myocardium without affecting perfusion to the same degree; septal:lateral glucose use ratio increased from 0.62 to 0.91 after CRT, p<0.001.39,93,94 Regarding the septum in particular, a slower – anticipated by the fact the lateral segments are also simultaneously actively contracting – yet more effective contraction has been reported.95 The importance of myocyte length has been further demonstrated by the dependence of generated pressure and oxygen consumption (both increasing, the former more than the latter, leading to improved efficiency) on atrioventricular delay during CRT, with extremely short delays completely negating haemodynamic effects of resynchronisation.96 Moreover, CRT appeared to confer a benefit with regard to the metabolic reserve and mechanical efficiency (response to beta-stimulation) of the failing heart, even after long-term application.72 The same increases in output were noted upon dobutamine administration compared with dyssynchronous hearts, yet were accompanied by increased efficiency and acetate extraction (operation at less-than-maximal oxidative capacity at rest).93 This long-term trend – present even 13 months after CRT initiation – alludes to intrinsic metabolism alterations after prolonged CRT application, consistent with the notion of detrimental effects of dyssynchrony itself on energy efficiency.72,97 Additionally, a trend for beneficial effects of CRT on fluid dynamics, leading to increased direct conversion of inflow to outflow kinetic energy, has been reported. This complements previous reports on the effects of dyssynchrony on rheodynamics and potentially offers a novel marker for predicting response to CRT.98–101

Although similar metabolism alterations have been reported in ischaemic cardiomyopathy and analogous CRT effects are indeed observed in the majority of dyssynchronous cases, a sizeable minority (32%) do not exhibit septal reverse mismatch (lower glucose use relative to perfusion).102 This is attributable to perfusion deficits of the lateral wall – present in 91% of the subgroup discussed – precluding higher glucose use there and, in effect, leaving the septum responsible for cardiac output.
An important inherent limitation of this approach for metabolism assessment lies in the assumption of intracellular homogeneity that ignores the high degree of compartmentalisation and sequestration in the sarcoplasm.9,103 That is, intracellular presence of a metabolite does not account for the site and mode of its breakdown, its vicinity to the energy-consuming areas of the cell and ultimately its energy yield, alterations of which may crucially affect efficiency.
A novel pacing modality, multisite pacing – constituting an advanced form of CRT – has started to yield interesting results regarding the previously mentioned parameters. It entails administration of two, rather than one, left ventricular pacing pulses, followed by a stimulus to the right ventricle. As such, it allows for sculpting of the activation sequence of the myocardium, potentially bypassing limitations posed by dense scar presence and allowing for further optimisation.104 Interesting results have been obtained regarding haemodynamics and stroke work improvement, and clinical studies attempting to correlate multisite pacing optimisation with energy efficiency improvement at the cardiovascular system level, through use of ventriculoarterial coupling, are underway.105,106
Dyssynchronous heart failure, specifically in the form of left bundle branch block presence, leads to mechanical dispersion and altered stretching of the different myocardial segments, affecting calcium ion homeostasis – potentially via mechanosensitive ion channels – leading to proarrhythmia, theoretically reversible by CRT.107,108 However, CRT can, itself, under certain conditions, exhibit proarrhythmic effects as a result of:109
- increased transmural dispersion of repolarisation (pulse delivered at left ventricular epicardium);
- wave break at the collision area of the two pulses (left and right ventricular) and potential re-entry; and
- in-scar pacing, amenable to time-dependent alteration, potentially underlying electrical storm events.110,111
As as result of the first pulse being delivered with increased width, multisite pacing may not be involved in re-entry occurring because of localised in-scar pacing.104
This implies that CRT, besides its acute haemodynamic effects – stroke volume increases were noted in the previously mentioned studies – has profound effects on myocardial energy management.72,93 This raises the intriguing possibility that, depending on currently undetermined individual parameters, response to CRT may not manifest as an overt improvement in ejection fraction, chamber volumes and pressures, but in covert alterations in metabolism, increasing efficiency and reserve, rendering the heart more able to respond to abrupt increases in output demand.112 Such effects have been reported even when classical adverse prognosticators – QRS widening upon biventricular pacing – are observed, suggesting a degree of independence between effects on mechanics and bioenergetics, owing to differing underlying molecular pathways.113 The inhomogeneous myocardial involvement in ischaemic cardiomyopathy, leading to divergent and unpredictable effects of dyssynchrony on metabolism, may also affect these covert responders, at least at the macroscopic (chamber) level.102
CRT Effects on Bioenergetics at the Ultramicroscopic Level
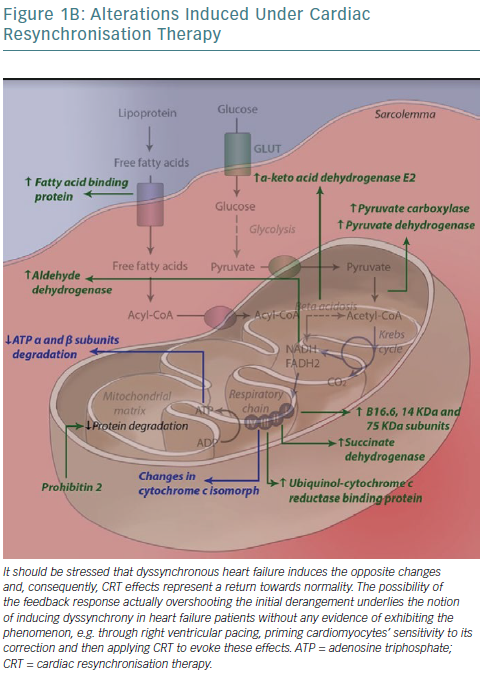
Mitochondria are one of the key sites of CRT effects at the ultramicroscopic level. Proteomic analysis in a canine model has revealed levels of 31 mitochondrial proteins to be altered after CRT application, with almost half constituting subunits of the respiratory chain.114 A concerted effect is observed whereby CRT increases activity of key enzymes in anaplerotic pathways, providing more intermediates of the Krebs cycle, which can otherwise be consumed to biosynthetic processes such as amino acid synthesis, increasing its activity, facilitating acetyl-coenzyme A entry into the cycle and consequently NADH/flavin adenine dinucleotide (FADH2) formation and funnelling of electrons into their transport chain to complete aerobic oxidation.115
Moreover, all complexes of the respiratory (electron transport) chain, with the exception of cytochrome c oxidase (complex IV), have critical subunits upregulated. This includes complex II (succinate dehydrogenase), which links the Krebs cycle with electron transport chain; it catalyses succinate to fumarate conversion along with FADH2 formation, immediately introduces FADH2-derived electrons into the chain and allows for the flow of NADH-derived electrons that enter the chain further upstream – complex I – as a result of higher energy levels towards complex III/cytochrome bc1 complex.116 Finally, ATP synthase (complex V) subunit degradation is inhibited and, following phosphorylation, complex formation and ATP yield per proton flowing is improved (2.5 ADP molecules phosphorylated per oxygen atom consumed with CRT versus 1.4 in dyssynchronous heart failure – due to electron entry now more often occurring at complex I). Complex V activity increases 20% during CRT compared with dyssynchronous heart failure. As such, ultimately, electrons exiting the chain and protons flowing down their electrochemical gradient through complex V are paired with molecular oxygen, forming water.
Several complementary effects have been noted, such as upregulation of fatty acid binding protein that allows their entry into mitochondrial matrix and subsequent beta-oxidation and downregulation of uncoupling proteins.117 The latter may be a defence mechanism, reducing energy production and – through substrate alteration – reducing contraction of the myocyte to prevent the occurrence of extreme stretches, especially during contraction against already shortened segments (lateral wall). Cytochrome c has, unexpectedly, been reported to be downregulated following biventricular pacing, owing to either methodological issues (post-translational modifications that alter its pI and so its localisation following protein electrophoresis) or in the context of preventing apoptotic cell death, promoted by several pathways in dyssynchronous heart failure, as previously discussed.114
Although electron and proton coupling with molecular oxygen is tightly controlled by complex IV, there is always the possibility of ROS formation through release of intermediates from complex IV or electron interaction with molecules other than oxygen. These can induce post-translational modifications to a multitude of mitochondrial energy production-related proteins and membrane lipids, severely impairing their functionality and even leading to cell death.118–122 Most importantly, endothelial nitric oxide synthase, under oxidative stress, becomes uncoupled from tetrahydrobiopterin (itself oxidated) and yields significant amounts of ROS.121
To negate this potentially detrimental side-effect of increased function and efficiency of oxidative metabolism, an increase in ROS-scavenging protein levels is necessary and sufficient. Indeed, CRT has been found to be linked to a significant rise in thioredoxin-dependent peroxide reductase, critical for the formation of reduced disulphide bonds that either constitute the reversal of oxidative events or may be coupled – as in the case of glutathione – to the reduction of a variety of oxidised molecules.114,123 Interestingly, post-translational modification of regulatory ATP synthase subunits (alpha subunits) has been reported (S-glutathionylation and S-nitrosation), along with novel disulphide bond formation. S-glutathionylation reversal by CRT was linked to a twofold increase in enzyme activity, providing a further mechanism for its effects on complex V.124 Despite this, downregulation of ATP synthase activity may be part of a feedback loop, where increases in oxidative stress, such as those caused by heart failure, prevent aerobic oxidation and ultimately limit ROS production.125–130 The ensuing increase in oxidative stress is alleviated by the aforementioned increases in ROS-scavenging proteins.
These changes in protein levels are coupled with increased expression of proteins that translocate into mitochondria – synthesis of most energy production-related proteins has been relinquished to the nucleus – chief among which is prohibitin 2. Prohibitins are assembled into a ring-like structure in the inner mitochondrial membrane and their presumed roles involve being chaperones for respiration chain proteins (ensuring proper protein folding) or acting as general structuring scaffolds required for optimal mitochondrial morphology and function.131,132 Mitochondrial protein proteases have conversely been found to be downregulated post-CRT.133
Of note, according to transcriptomic analyses, gene level-related effects of dyssynchrony were sixfold more pronounced at the anterior than the lateral wall.134 Appropriately, CRT leads to pronounced local but not global changes in gene expression.26 Chronically increased stresses such as those observed upon dyssynchrony lead to profound alterations in key elements of the contractile apparatus, namely the transverse tubules (T-tubules), causing their disruption. This is mediated by downregulation of structural proteins such as junctophilin, and consequently impairs calcium handling (T-tubules bring into proximity the sarcolemmal and SR membranes).135,136 This indirectly affects energy use as much larger (potentially proarrhythmic) calcium spikes are necessary to cause calcium release from the SR and accomplish the excitation-contraction coupling; a disturbance that CRT can rectify.
A potential master regulator of protein activity affected by CRT has been recognised in the form of CK2 (formerly casein kinase II), a serine-threonine protein kinase, using both ATP and guanosine-5’-triphosphate as substrates and indispensable to cell survival and growth by preventing caspase access to cleavage sites of proteins, promoting DNA repair and suppressing p53 activity.137–139 In a canine model of tachypacing- and left bundle branch-related dyssynchronous heart failure, phosphoproteomics analysis identified CK2 as the most likely kinase whose downregulation is involved in protein phosphorylation alterations observed in dyssynchronous heart failure, a pattern reversed by the introduction of CRT. Although an intriguing finding, it cannot be inferred whether CRT effects are mediated by upregulation of CK2 or whether improved energetics and triphosphate nucleotide availability (used by CK2 as a phosphate donor during phosphorylation) account for increased kinase activity, promoting cell survival and function in a virtuous cycle. Finally, similar effects leading to increases in oxidative potential have been noted in peripheral muscles as well, potentially resulting in further improvement of patients’ functional status and exercise tolerance.140
Increased adrenergic signalling is usually thought of as deleterious in heart failure; evidenced by the benefits of its blockade with beta-blockers. Accordingly, there is intrinsically reduced responsiveness of the myocardium to adrenergic stimuli as a defence mechanism involving increased activation of inhibitory G-alpha subunits (deactivating rather than activating protein kinase A; PKA) and internalisation/degradation of receptors through phosphorylation by G-protein receptor kinase 2.141–144 However, the response of the myocardium to a transient increase in adrenergic signalling may be crucial in maintaining exercise capacity. CRT has been found to restore this parameter by upregulating regulator proteins of G-protein signalling (RGS) that inhibit inhibitory alpha-subunits of G-proteins (Gais).145 As such, SR-bound PKA may activate calcium-handling proteins, increasing calcium ion transient flux during depolarisation and its subsequent sequestration, ultimately enhancing the associated myocyte shortening, reversing the severely blunted calcium spikes of dyssynchronous heart failure.146–148 This post-translational effect on calcium-handling proteins is supported by the observation that their encoding genes are not upregulated by CRT.149 Consequently, exercise tolerance may be further improved.
The most intriguing observation stemming from a canine model study by Chakir et al. lies in the triggering of increased response to beta-adrenergic signalling following dyssynchrony introduction into synchronous heart failure.145 This allowed for radical hypotheses claiming that a CRT holiday (analogous to diuretic holiday) may enhance CRT effects – particularly in non-responders – and that purposeful transient dyssynchrony introduction in heart failure patients without an indication for CRT or a pacemaker may trigger the same beneficial results. Animal studies in the tachypacing-induced cardiomyopathy setting have since confirmed this assumption, reporting improved cardiac response to beta-adrenergic stimuli and suppression of heart failure in terms of chamber dilatation and cellular dysfunction, even in the context of synchronous heart failure.150 An obvious noted issue stems from potential intersubject differences in the required holiday period.
A further path linking substrate preference to contractile function has been reported to be affected by CRT, namely phosphorylation of Z-disk and M-band proteins by the upregulated glycogen synthase kinase.151 Activation of this kinase leads to inactivation of glycogen synthase, potentially in the context of restoring the omnivorous nature of the cardiomyocyte. Target sarcomeric proteins (troponins I and T, myosin-binding protein C and myosin light chain isoforms) are involved in calcium sensing and consequently their activation sensitises the contractile apparatus to calcium ions’ presence and facilitates contraction.
Moreover, CRT has been shown to increase messenger RNA levels of the alpha-myosin isoform and its relative ratio to those of beta-myosin in advanced dyssynchronous heart failure patients, possibly because of improvements in energy metabolism that allow for a more ATP-consuming yet more efficient isoform to be chosen, restoring functionality.152 Interestingly, another subset of glycogen synthase kinase target sarcomeric proteins are involved in mechanosensing and may actually partake in the core mechanisms through which CRT exerts its effects. Figure 1 summarises the most important effects of CRT application to metabolism in dyssynchronous heart failure at the ultramicroscopic level.
Linking Mechanics to Metabolomics
A fundamental issue regarding all the described effects of CRT on cellular bioenergetics is raised concerning mechanisms and signal transduction pathways underlying the translation of mechanical alterations to metabolic effects. A strong candidate is the cytoskeleton, linking sarcomeres – the contractile units of cardiomyocytes – with the extracellular matrix and ensuring that cellular contraction causes tissue shortening. As a result of changes in myocardial lamellae orientation and chamber architecture, mechanical deformation of the failing heart is significantly altered.153,154 The aforementioned changes in calcium concentration, owing to alterations in proteins such as SERCA, decrease peak contractile force at the same time as modified extracellular matrix synthesis (different collagen isoforms) and sarcomere architecture (reduced levels of titin, a spring-like protein conferring elasticity) render myocardial tissue stiff.155,156
Concomitantly, changes in gap junction proteins – connexin-43 is redistributed laterally rather than longitudinally – reduce both conduction velocity and synchronisation between adjacent cardiomyocytes.157,158 Protein kinase B, which is upregulated by CRT, has been found to phosphorylate connexin-43 and facilitate incorporation into gap junctions, allowing for initiation and coordination of the ischaemic injury response.21,159 Consequently, mitogen-activated protein kinase further phosphorylates connexin-43 leading to gap junction closure and severing communication between adjacent cells’ sarcoplasms, potentially preventing apoptosis spreading.159 As such, stiff cardiomyocytes exhibiting reduced functionality attempt to contract over a stiffer matrix, not amenable to deformation, leading to abnormally high tension to the cytoskeletal structures connecting them (focal adhesions and integrins).160 This condition is further aggravated by local (between adjacent cells) and regional (in dyssynchronous heart failure) lack of coordination.156,161
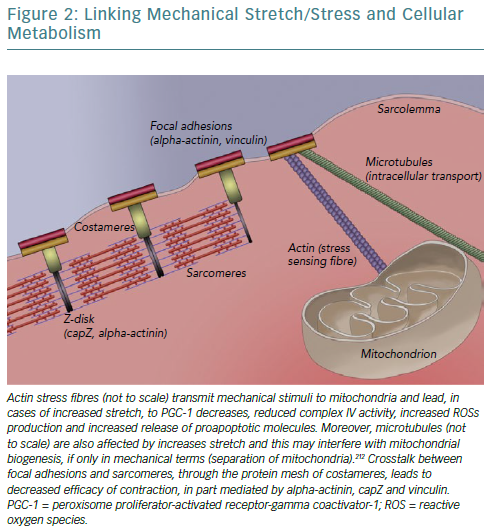
Focal adhesions are so intricately linked with sarcomeres – to ensure anchoring to sarcolemma/extracellular matrix and bidirectional mechanotransduction – that a specialised structure emerges called a costamere.162–164 Costameres are sub-sarcolemmal multiprotein complexes initially found to localise over Z-disks, perpendicularly to the cardiomyocyte axis, but subsequently shown to extend over M-lines and along the myocyte length.163,165,166 They can be thought of as a specialised striated and cardiac muscle version of focal adhesions. Two main constituent protein complexes have been identified; the dystrophin/glycoprotein complex that provides a link between laminin – an extracellular matrix protein – and filamentous actin-based cytoskeleton, and the integrin/talin/vinculin complex, which is potentially more involved in signal transduction given its association with integrin-linked and focal adhesion kinases.167–170
The importance of these protein complexes in the biomechanical stability of the heart, especially under mechanical stress, can be inferred by the fact that mice with cardiac-specific vinculin deletion display a dilated cardiomyopathy phenotype.171 Moreover, another integrin-associated protein, melusin – a chaperone, assisting in proper protein folding and assembly – performs a highly specialised role in cardiomyocyte response to stress stimuli by promoting cellular survival and hypertrophy through both mitogen-activated protein kinase family pathways and protein kinase B (also affecting energy metabolism and, as mentioned previously, upregulated by CRT).58,59,172–174 Indeed, in aortic stenosis patients, melusin levels have been found to correlate with systolic function preservation.175 As such, it could be theorised that persistent supraphysiological stress (stretch) levels, such as those in dyssynchronous heart failure, render melusin levels insufficient to induce and maintain proper cellular responses, leading to apoptosis and myocardial wall thinning.
Similar findings have been reported in the case of integrins.176 Changes in integrin isoform gene expression profile have been noted during reverse remodelling of chambers upon left ventricular assist device therapy.176 CRT has been shown to lead to alterations in levels or activity of several sarcomeric and focal adhesions-related proteins involved in mechanosensing, such as cap-Z (reduced expression), muscle LIM protein (acetylation-activation), tensin, desmin and filamin-C.151,177–180 Spatial distribution of alpha-actinin – a microfilament protein necessary for the attachment of actin filaments to the Z-lines in skeletal muscle cells, coordinating sarcomeric contraction and providing links to focal adhesions and stress actin fibres – has been found to be disrupted in dyssynchronous heart failure, losing its periodicity and forming depositions, with CRT partially rectifying its pattern.181–183
Mitochondria, being linked to the cytoskeleton – both actin and microtubules – for reasons of localisation and transport, also sense the increased tension of dyssynchronous heart failure.55,184,185 This leads to increased expression of proapoptotic Bcl-2 family proteins, including Bax and Bad, which mediate cytochrome c release and apoptosis triggering, and to reduced complex IV activity (reduced by 21%) leading to reduced ATP formation and ROS accumulation because electrons are unable to combine with molecular oxygen and protons.60,186,187 In fact, the type of stretch caused by dyssynchrony (monotonous stretch) further accentuates these changes and directly reduces ATP synthase activity and mitochondrial biogenesis by reducing PGC-1alpha.188 In this framework, shutting down oxidative metabolism – with all its detrimental consequences – may again constitute a defensive mechanism of the cell to prevent apoptosis, in part caused by increased mechanical stretch. As such, not only is metabolism affected by anomalous stretch, but also its ability to adapt in response to such stressors is impaired.189
As such, an overarching hypothesis could be that alleviation by resynchronisation of the increased strain imposed on cardiomyocytes by dyssynchrony, which led to compensatory alterations/remodelling of sarcomeres and triggered changes in bioenergetics, now leads to altered input from specialised mechanosensors that is transduced to the cell, triggering the beneficial effects.53,190 A depiction of our current partial understanding of this link is attempted in Figure 2.
Conclusion
CRT can be thought of as constituting a metabolic therapy, acting on two levels. Firstly, at the chamber level, it rehomogenises substrate use and improving mechanical output and energy efficiency by reducing energy spent on internal work. Secondly, at the cellular level, it increases oxidative cell capacity by acting at virtually all stages of oxidative metabolism. The latter effect may not be inseparable from the former, creating the potential for the existence of covert responders.
Whether this is true for cases that do not have an indication for CRT or can be achieved through more advanced resynchronisation modalities, such as multisite pacing, or even by allowing (in dyssynchronous heart failure) or introducing (in synchronous heart failure) intermittent dyssynchrony, requires further study. It is possible that advanced computational models will be needed to determine the optimal site of leads and timing parameters to achieve conventional CRT/multisite pacing optimisation. Certain models can already integrate projected changes in bioenergetics, in the form of ATP use homogenisation assessed concomitantly with stroke work maximisation, to propose adequate lead implantation sites.156,191 Although challenging, especially to the clinician, comprehending these principles will probably prove necessary to promote our insight into the effects of resynchronisation beyond chamber mechanics, improving care for our patients.