







Amyloidosis is a condition characterised by accumulation of pathologic fibrillar proteins in organs causing dysfunction.1 Several protein precursors have been shown to cause amyloidosis.2 Cardiac amyloidosis, when waxy, starch-like deposits infiltrate the heart, is most commonly secondary to the accumulation of amyloid fibrils derived from immunoglobulin light chains (AL) or transthyretin (ATTR). Most of the early literature regarding cardiac amyloidosis investigated the AL type, which is often associated with extra-cardiac manifestations with multi-organ involvement. The Mayo staging criteria, as reported by Dispenzeiri et al., are used to grade systemic AL amyloidosis associated with plasma cell dyscrasias. Prognosis in AL amyloidosis correlates with cardiac biomarkers troponin and brain natriuretic peptide (BNP), regardless of confirmed cardiac involvement by routine assessment.3 Cardiac AL amyloidosis has poor prognosis, with most studies reporting median survival of 6–12 months from the date of diagnosis.4,5
More recently, the ATTR subtype has been identified as an important cause of cardiac amyloidosis.6 ATTR amyloidosis is further subdivided into senile cardiac amyloidosis, due to amyloid fibrils composed of wild-type non-mutant transthyretin (ATTRwt), and hereditary forms caused by gene mutations in the transthyretin gene on chromosome 18 (ATTRm). Overall prognosis for cardiac ATTR amyloidosis is better than for the AL type, with median survival typically 2–6 years.7–9
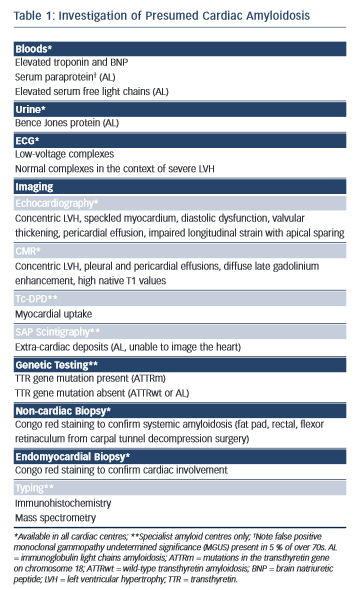
Amyloidosis remains a histological diagnosis, with confirmation requiring microscopic examination of amorphous material in affected tissue in which Congo red displays apple-green birefringence when viewed under high-intensity cross-polarised light.10 Immunohistochemical staining of the amyloid deposits is performed using the peroxidise anti-peroxidase method to confirm the amyloid fibril type, using purified immunoglobulin G (IgG) fractions of monospecific antibodies reactive with serum amyloid A protein (SAA), transthyretin (TTR) and with kappa and lambda immunoglobulin light chains.11 Laser microdissection and proteomic analysis using mass spectrometry is now considered the gold standard test to identify the amyloid type but is not widely available.12 A summary of useful investigations in patients with presumed cardiac amyloidosis can be found in Table 1 with further explanation throughout the text.
Pathophysiology
There are data to suggest prognosis in AL amyloidosis is poor due to myocardial light chain toxicity.13 Pathophysiology due to ATTR amyloidosis is probably related to infiltration alone, with concomitant cell hypertrophy contributing to increased wall thickening.14 New insights into the pathogenic processes involved in ATTR amyloidosis have been reported. Transthyretin, a transporter protein for thyroxine and retinol in the blood, is formed from four subunits.15 Instability of the tetrameric transthyretin protein has been shown to increase the likelihood of forming amyloid fibrils.16 Proteostasis in all ATTR types may be as important as the point mutations causing ATTRm, as evidenced by overexpression of extracellular chaperones in ATTR patients.17 Proteolytic cleavage has been identified as an important step in destabilisation of the TTR tetramer. The mechano-enzymatic mechanism described by Marcoux et al. is common to several amyloidogenic variants and is particularly important in the heart where shear stress, a process increasing susceptibility to proteolytic cleavage, is greatest.18 The findings are encouraging for the development of therapies targeting inhibition of transthyretin amyloidogenesis.
The Importance of ATTR Cardiac Amyloidosis
Two separate post-mortem series reported a high incidence (25 %) of transthyretin amyloid deposits in the myocardium of elderly subjects at the time of death.19,20 It is not yet known whether the presence of transthyretin amyloid in the heart of very elderly subjects is part of a general aging process or represents under-diagnosis of the clinical phenotype pre-mortem. ATTRwt is associated with an isolated cardiac phenotype, presenting as diastolic heart failure in the eighth decade, with approximately 9:1 male:female ratio.7 Wildtype ATTR amyloidosis is increasingly recognised as an important, yet often unrecognised, cause of heart failure with preserved ejection fraction (HFpEF), a likely heterogeneous group with differing aetiologies. Nuclear imaging with technetium-99m-labelled (99mTc) 3,3-diphosphono-1,2-propanodicarboxylic acid (Tc-DPD) was used to screen 120 HFpEF patients and 16 patients (13.3 %) showed significant cardiac uptake, later confirmed as ATTRwt following genetic testing and endomyocardial biopsy.21 Unlike the vast majority of studies on ATTRwt, there was no difference in sex in the HFpEF patients with cardiac amyloidosis, suggesting the diagnosis by other methods is often missed in women.22 Undiagnosed ATTRwt has been suggested as a possible cause of poor outcomes in patients undergoing transcutaneous aortic valve replacement (TAVR).23 Coexistent degenerative aortic stenosis and ATTRwt has been reported in a small series of 5/43 patients undergoing aortic valve replacement, with confirmed wild-type TTR on endomyocardial biopsy.24 The authors propose larger prospective studies to determine the role of screening for cardiac amyloidosis with Tc-DPD to aid risk stratification in patients being selected for aortic valve interventions.
Over 100 TTR gene mutations have been associated with systemic amyloidosis, a condition with autosomal dominant inheritance.25 The V122I variant, in which valine is substituted for isoleucine at position 122, has been the focus of significant interest in recent years. V122I amyloidosis was first reported in 1989 and the clinical phenotype is identical to ATTRwt, with isolated cardiac involvement.26,27 V122I allele frequency was first reported as 3.9 % in African Americans in 1996 and has more recently been re-calculated in a study analysing DNA from 1688 New York State African American newborns.28,29 TTR V122I was detected in 65/3376 alleles and, through expansion of the analysis to include samples from a ‘wellness’ study in San Diego, the authors calculated 3.43 % of African Americans under 65 carry at least one copy of the amyloidogenic allele. Disease onset is reported from 65 years and thus, with the aging population, and improving detection techniques, ATTR V122I is expected to be confirmed as the cause of heart failure in increasing numbers of elderly black patients. In January 2015, it was reported by Quarta et al. in The New England Journal of Medicine that there was no significant difference in mortality for V122I carriers and the prevalence of overt cardiac abnormalities was low, following an interim analysis of the Atherosclerosis Risk in the Communities (ARIC) study.30 Final follow-up, totalling 21.5 years, took place before the median age of presentation with ATTR V122I.31,32 As a result, the authors may have underestimated the true burden of cardiac amyloidosis due to ATTR V122I.33
Imaging
Echocardiography
Echocardiography is generally the first imaging modality in the investigation of cardiac disease; it is widely available and can be performed at the bedside. Echocardiography is well established in the diagnosis of cardiac amyloidosis with characteristic features. Increased, often concentric, wall thickening with a bright ‘speckled’ appearance of the myocardium is a characteristic finding, often misinterpreted as left ventricular ‘hypertrophy’ despite a different underlying process to hypertrophic cardiomyopathy or hypertensive heart disease. Valvular thickening, diastolic and systolic dysfunction, abnormal strain with typical apical-sparing and pericardial effusion are features suggesting amyloid infiltration.9,34–36 Most studies report increased wall thickening in ATTR compared with AL amyloidosis but longitudinal strain impairment is similar and does have adequate sensitivity or specificity to distinguish between types. A recent small study found no statistically significant echocardiographic differences between cardiac AL and ATTR amyloidosis when comparing systolic and diastolic function but distinguished the types using a ratio of BNP and left ventricular mass index that was higher in AL patients.37 Echocardiography has been used in combination with ECG to detect early cardiac involvement in ATTR amyloidosis. Low-voltage ECG complexes are frequently seen in AL (71 %) and ATTR (56 %) but with poor sensitivity.38,39 The combination of low-voltage ECG and septal wall thickness >14 mm can help to identify early cardiac amyloidosis in subjects with transthyretin gene mutations,40 but nuclear imaging is now considered the gold standard for preemptive screening.
Cardiovascular Magnetic Resonance
Cardiovascular magnetic resonance (CMR) produces high-resolution images of the heart through complex physics and computational algorithms by using the magnetising properties of atomic nuclei in tissues. Delayed images are routinely acquired after gadolinium infusion, an intravascular contrast agent known to accumulate in regions of extracellular expansion. Several studies have shown the high sensitivity of late gadolinium imaging in cardiac amyloidosis and the findings have been correlated with histological examination of explanted tissue.41–43 More recently, novel T1 mapping techniques to interrogate the native tissue characteristics and quantify the extracellular volume have been deployed in the investigation of cardiac amyloidosis.44,45 A relatively large study of the prognostic value of late gadolinium enhancement (LGE) CMR has been published recently by Fontana et al.46 The authors highlight the additional benefit of phase sensitive inversion recovery (PSIR) imaging to eliminate potential operator-dependent errors relating to the abnormal gadolinium kinetics observed in cardiac amyloidosis41 and no difference in the pattern of LGE was found between the amyloid subtypes. There are conflicting reports in the literature regarding the differentiation of amyloid types by deposition patterns, including a study by the present author.47 A recent histological study of postmortem specimens found diffuse pericellular deposits suggesting AL and nodular deposits more commonly observed in ATTR, from which it can be inferred that morphological differences may be observed according to amyloid fibril composition.48
Nuclear Imaging
Nuclear cardiac imaging has emerged as an imaging modality with high sensitivity in the detection of cardiac amyloid deposits, particularly the ATTR subtype.49 Several phosphonate-based tracers, including 99mTcpyrophosphate (Tc-PYP) and Tc-DPD, have been reported to localise amyloid in the heart as a separate property to their original use in bone scintigraphy.50 There are no reports of false negative Tc-DPD scans in patients with confirmed cardiac ATTR amyloidosis and more than half of patients with cardiac AL type have positive scans.51 Tc-DPD is being considered as a screening investigation to rule out cardiac involvement in carriers of ATTR mutations since positive results may occur before abnormalities on echocardiogram.52 Nuclear imaging can be performed in patients who have contraindications to cardiac MRI, such as non-compatible pacing devices or implantable cardioverter defibrillators (ICDs).
Cardiac Computed Tomography
Extracellular volume quantification is well established in the assessment of cardiac amyloidosis by cardiac MRI techniques (equilibrium contrast CMR [EQ-CMR]). A recent study has reported equilibrium cardiac computed tomography (CT) using pre- and postcontrast calculation of myocardial volume, with good correlation compared with EQ-CMR and using a shorter protocol.53 Cardiac CT may be a useful alternative to CMR for patients with contraindications in whom quantification of amyloid burden is important for serial monitoring. Ionising radiation is unlikely to be a major concern in the more elderly patients, particularly in the context of a condition with relatively poor prognosis, and doses are lower than in conventionally used bone tracer scintigraphy.
Treatments
Correct diagnosis of cardiac amyloidosis is important to identify patients requiring specialist treatments that are not widely available, and to avoid potentially harmful but otherwise routine heart failure treatments. Cardiac amyloidosis patients are often unresponsive to diuretics at normal dosage and there are no data to support the prognostic benefit of angiotensin converter enzyme (ACE) inhibitors. Calcium-channel blockers and digoxin are potentially toxic.6 The high incidence of progressive heart block in some amyloid subtypes suggests a relative contraindication to beta-blocker use, at least in the absence of regular ECG monitoring.39
Chemotherapy
Chemotherapy is not indicated in ATTR amyloidosis and correct identification of amyloid type is necessary to prevent inappropriate chemotherapy for presumed systemic AL amyloidosis. Lachmann et al. investigated 350 patients with a diagnosis of apparent AL amyloidosis and found an alternative genetic cause in 9.7 %, including about 5 % patients with hereditary ATTR amyloidosis.54 The presence of a monoclonal gammopathy had often been misleading in this series, suggesting AL amyloidosis, but it is important to appreciate that incidental monoclonal gammopathy undetermined significance (MGUS) occurs in over 5% of persons aged over 70 years.55
Chemotherapy regimes in cardiac AL amyloidosis aim to suppress the production of amyloidogenic immunoglobulin light chains produced by plasma cell clones in the bone marrow. Combination therapy including an alkylating agent, such as melphalan or cyclophosphamide, and a steroid is the backbone of systemic AL amyloidosis treatment.56 The proteasome inhibitor bortezomib (Velcade®) has advanced the treatment options and when used in combination with cyclophosphamide and dexamethasone (CVD) resulted in good haematological response rates of 81 % in a study of AL patients of whom 74 % had cardiac involvement.57 CVD is not well tolerated in advanced cardiac amyloidosis and patients must be monitored closely during the first cycles, when cardiac decompensation may occur. Patients with good early response, identified by a rapid reduction in serum free light chains, troponin and BNP, have the best outcomes.56
Transplantation
Organ transplantation is a potential cure for amyloidosis. Availability of donor hearts is low in the UK; advanced age and multi-organ failure further precludes cardiac transplantation in many patients. Autologous stem cell transplantation in AL amyloidosis is recommended following cardiac transplantation to remove the pathological plasma cells, adding to overall risk, with mortality 2–10 % in patients with cardiac or multiorgan involvement.56 The liver produces 95 % of serum TTR and thus liver transplantation may be necessary in addition to cardiac transplantation in ATTRm but is not indicated in ATTRwt given the on-going production of non-mutant TTR. Progression of cardiac amyloidosis following orthotopic liver transplantation has occurred in all patients with the T60A variant treated at the National Amyloidosis Centre.8
A recent single-centre study reported the results of cardiac transplantation in nine patients with AL amyloidosis and 10 patients with ATTR amyloidosis.58 The median age of patients was 59 years and there were two deaths over 380-day follow-up. One-year survival was 100 % for AL patients and 74 % for ATTR patients. The authors attribute the good outcome in AL patients to aggressive control of light chains; eight out of nine patients received chemotherapy prior to transplantation and five patients underwent autologous stem cell transplantation.
Novel TTR Treatments
ATTR amyloidosis has emerged as an exciting area of therapeutic development.59 Treatment strategies target three areas: inhibition of hepatic TTR production; TTR stabilisation; and increased clearance of TTR amyloid fibrils.60
TTR Inhibition
One hypothesis is reducing TTR production will ultimately lead to less amyloid deposition, allowing the normal turnover to exceed accumulation of amyloids in organs and tissues. The small interfering RNAs (siRNAs) that are bound to RNA-inducing silencing complexes mediate cleavage of target messenger RNA (mRNA). The siRNA agent ALN-TTRSC (Alnylam Pharmaceuticals, Cambridge, MA) has been shown to markedly suppress TTR production in ATTR patients and healthy volunteers61 and a phase III clinical trial in familial amyloid cardiomyopathy is in progress. Antisense oligonucleotides (ASOs) are synthetic single-stranded oligomers designed to bind to specific regions of target mRNA. A phase III study of the ASO agent targeted to TTR mRNA, ISIS-TTRRx (Isis Pharmaceuticals, Carlsbad, CA), is in progress in patients with familial amyloid polyneuropathy (FAP).
TTR Stabilisers
Tafamadis (Vyndaqel®) is a small molecule that selectively binds to the thyroxine binding site of TTR in plasma and stabilises the tetrameric protein in vitro.62 Tafamadis has been shown to reduce the rate of change in peripheral neuropathy scores in FAP and the encouraging results have prompted a phase III clinical trial in ATTR cardiomyopathy.
Diflunisal is a non-steroidal anti-inflammatory drug that binds to the thyroxine binding sites on the TTR tetramer.16 Diflunisal is associated with the typical non-steroidal anti-inflammatory (NSAID) related adverse events, including gastrointestinal bleeding, renal dysfunction and worsening congestive heart failure, all of which may have devastating consequences in patients with amyloidosis. The results of a randomised, double-blind placebo-controlled trial to assess diflunisal in peripheral and autonomic neuropathy showed a reduction in the rate of progression of neuropathy compared with the placebo.63 Diflunisal has been assessed in a single-arm study involving a small number of cardiac ATTR amyloidosis patients (n=13) in whom tolerability was confirmed but the drug has not been investigated in a randomised trial.64 The use of diflunisal remains an off-label indication in the UK and should only be prescribed in specialist amyloidosis centres with the relevant licence.
Increased Clearance of TTR Amyloid Fibrils
The antibiotic doxycycline has been shown to disrupt amyloid fibrils in a transgenic mouse model.60 A phase II open label study investigating doxycycline in 20 FAP patients over 12 months showed no clinical progression of cardiac involvement and stable neuropathy scoring.65 Further investigation of doxycycline in ATTR amyloidosis is needed to assess the role in the wider patient population.
Serum amyloid P component (SAP) is present in all types of amyloid. The drug (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine- 2-carboxylic acid (CPHPC) is a novel small molecule drug that crosslinks SAP in the plasma, triggering hepatic depletion.66 The combination of CPHPC and anti-SAP antibodies has been shown to eliminate visceral amyloid deposits in mice.67 This immunotherapeutic approach to treatment has been confirmed as safe in man with a recently reported phase 1 trial involving 15 patients with systemic amyloidosis.68 Further studies are needed to confirm the role of the potential cure for systemic amyloidosis in patients with cardiac involvement.
Conclusion
Cardiac amyloidosis, previously considered a rare condition, is becoming an increasingly important cause of heart failure. Progressive imaging techniques have markedly increased detection rates. The ATTR type in particular is likely underdiagnosed in elderly and Afro-Caribbean populations. Treatment options have been limited until recently but several therapeutic strategies are currently undergoing phase III clinical trial assessment. Many more patients with cardiac amyloidosis will be identified with increased awareness and should be referred to specialist centres for further characterisation, implementation of an appropriate management plan and allowed access to novel drugs.