







Acute coronary syndrome (ACS) is a term used to encompass unstable angina (UA) and myocardial infarction (MI) with or without electrocardiographic (ECG) evidence of ST-segment elevation. Antiplatelet therapy has formed the backbone of ACS management for decades and the drug class continues to evolve as novel agents with increasingly efficacious antiplatelet actions are identified. The main risk associated with all antiplatelet therapies is bleeding, and physicians need to carefully weigh the possible adverse effects against the benefits of prescribing these drugs to patients with ACS. Aspirin, which has been recognised to have antithrombotic effects since the 1960s, continues to be prescribed almost ubiquitously for patients with ACS, and P2Y12 antagonists are now often added in; such dual antiplatelet therapy confers greater antithrombotic efficacy but at the risk of increased bleeding. Over recent years, it has become apparent that these drugs may also exert powerful anti-inflammatory effects that provide additional benefit in the management of ACS. This review will discuss antiplatelet agents that are currently used to treat patients with UA and MI.
Aspirin in Acute Coronary Syndrome
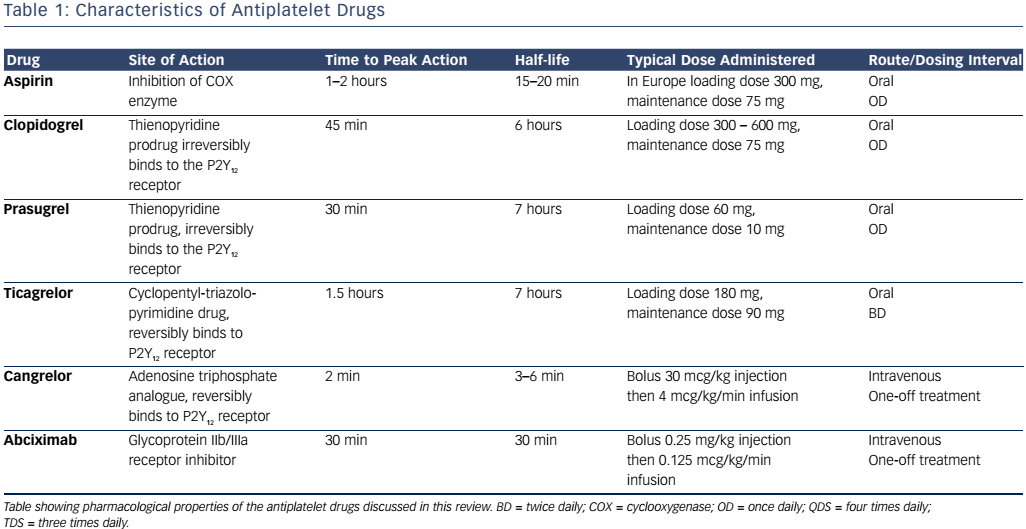
Alongside its analgesic and antipyretic effects, aspirin has been recognised as an inhibitor of platelet function for around 50 years1 and, to date, remains the most commonly prescribed drug worldwide.2 Current guidelines for the treatment of ACS recommend that all patients routinely receive a loading dose of aspirin, followed by maintenance therapy unless contraindicated.3,4 There is a wide range of maintenance dosages of aspirin that are prescribed in this context, ranging from 75 to 325 mg daily in different countries.5 In patients who have suffered a MI, recommended secondary prevention therapy typically entails 12 months of dual antiplatelet therapy, followed by lifelong aspirin, whilst patients with angina (stable or unstable) commence aspirin monotherapy.6,7
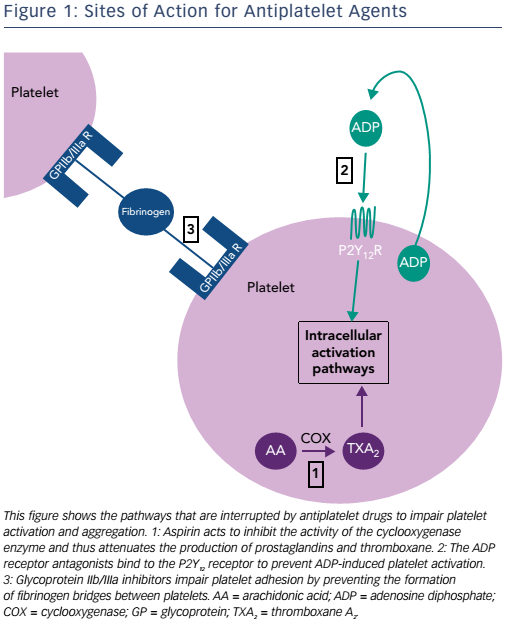
The cyclooxygenase (COX) enzyme converts arachidonic acid to prostaglandins. There are two human isoforms: COX-1, which is expressed in most tissues and COX-2, which is typically present in areas of active inflammation but can also (like COX-1) be expressed constitutively in some tissues.8 Prostaglandins are a large subgroup of eicosanoids; of these, two derivatives, prostacyclin and thromboxane A2 (TXA2), are of particular importance in maintaining platelet homeostasis. Prostacyclin inhibits while TXA2 increases platelet activation. Aspirin irreversibly inhibits both COX enzymes through acetylation of a critical serine residue, although its effect on COX-1 is at least tenfold greater than that on COX-2.8 By predominantly inhibiting COX-1, TXA2- induced platelet activation and aggregation are blocked.9 Circulating platelets have a lifespan of 8–10 days, and are typically present at a concentration of 150–400 x 109 per litre of blood (see Figure 1 and Table 1).10 Aspirin achieves maximum platelet inhibition within 2 hours following a loading dose and inhibits aggregation in 50 % of circulating platelets for at least five days.11,12 There has been some debate over the past decade as to whether aspirin resistance is a clinically significant issue, requiring patients deemed to be ‘poor responders’ to switch to an alternative antiplatelet agent.13,14 It has since been shown that whilst platelet function assays may produce variable results, and thus suggest that aspirin resistance is present, platelet COX-1 activity, as reflected by TXA2 levels, is uniformly and persistently suppressed by low-dose aspirin15 and it is now generally accepted that true pharmacological resistance to aspirin is rare.16
Data from one of the first large-scale randomised controlled studies assessing the effectiveness of aspirin as an antithrombotic agent were published in 1983 – 1,266 men with UA received treatment with either aspirin or placebo, with results showing a significant reduction in progression to MI among those taking aspirin.17 Aspirin in the setting of acute MI has subsequently been found to have major beneficial effects on morbidity and mortality,18,19 and multiple meta-analyses have confirmed the pivotal role that aspirin plays in reducing cardiovascular events and mortality in patients with ACS.20,21
Adenosine Diphosphate Receptor Antagonists in Acute Coronary Syndrome
Alongside aspirin therapy, patients with UA or non-ST-elevation MI (NSTEMI) who are considered to have a predicted 6-month mortality of >1.5 % (which is the case for the vast majority of such patients), are typically treated with a loading dose of 300 mg clopidogrel.22 Those with acute ST-elevation MIs (STEMIs) will go on to receive coronary reperfusion therapy, usually with percutaneous coronary intervention (PCI) but more rarely these days with fibrinolytic therapy, and also receive a second antiplatelet agent, namely clopidogrel, prasugrel or ticagrelor depending on local guidelines.
Clopidogrel, prasugrel and ticagrelor are all adenosine diphosphate (ADP) antagonists, a class of therapeutic agents that bind selectively to the P2Y12 receptor to inhibit platelet function (see Figure 1 and Table 1).23 The thienopyridine, ticlopidine was the first drug in this class but is seldom prescribed now, following reports of serious adverse reactions, in particular neutropaenia24 and thrombotic thrombocytopaenic purpura.25
Clopidogrel
The second generation thienopyridine prodrug, clopidogrel, is currently the most commonly prescribed ADP receptor antagonist. It is administered orally and up to 85 % of the absorbed drug undergoes hepatic metabolism by carboxylesterases to form an inactive carboxylic acid derivative, clopidogrelic acid, whilst the remaining 15 % is metabolised into the active thiol product by cytochrome P450 isoenzymes.23,26 Although clopidogrel has a relatively short half-life of 6 hours,27 the thiol metabolite covalently binds to the P2Y12 receptor, inducing an irreversible conformational change in the receptor and thus impairing thrombotic function for the remaining lifespan of the affected platelet. Genetic polymorphisms in cytochrome P450 enzymes, particularly CYP2C19 and CYP2C9, may result in impaired generation of the active thiol metabolite in patients taking clopidogrel, resulting in lack of efficacy.28 Variable responses to clopidogrel may additionally result from mutations in the ABCB1 gene that encodes the P-glycoprotein involved in clopidogrel absorption.28
The standard dosing regime is a 300 mg loading dose and 75 mg daily maintenance dose. Following this, steady state ADP inhibition is typically achieved within 3–7 days, with a 40–60 % reduction in ADPinduced platelet aggregation from baseline.27 Randomised controlled trials have demonstrated that clopidogrel is more effective than aspirin in preventing cardiovascular events in patients with vascular disease,29 reduces mortality further in patients with MI when used alongside aspirin,30 and improves outcomes in patients undergoing PCI again when used in combination with aspirin.31,32 The Clopidogrel versus Aspirin in Patients at Risk of Ischaemic Events (CAPRIE) study showed that clopidogrel administration was associated with similar adverse effects to those observed with aspirin, including gastrointestinal discomfort and increased bleeding, but the overall safety profile of clopidogrel 75 mg daily was considered to be at least as good as that of aspirin 325 mg daily.29 However, the inter-patient unpredictability in clopidogrel responsiveness (with some patients not responding at all) due to the above factors led to the development of newer P2Y12 antagonists.
Prasugrel
Prasugrel is an oral thienopyridine prodrug that is hydrolysed by esterases to the metabolite, R-95913. This inactive metabolite is then activated by cytochrome P450 enzymes, forming the active metabolite R-138727. As with clopidogrel, the active metabolite subsequently binds irreversibly via a covalent bond to the platelet P2Y12 receptor and thus inhibits platelet function.33
A loading dose of 60 mg is given, followed by 5–10 mg daily maintenance dosing.34 Peak plasma concentration is reached within 30 minutes and the drug has a half-life of 7 hours.33 Phase I and II studies have demonstrated that prasugrel has a faster onset of action than clopidogrel, as well as being more efficacious and more predictable in its antiplatelet action.35–37 The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 phase III study found that in patients with ACS undergoing PCI, prasugrel was more effective than clopidogrel in reducing further ischaemic events although it conveyed a higher risk of major bleeding.38 Further analysis of a subgroup of patients with STEMI undergoing PCI found that prasugrel was more effective than clopidogrel in preventing additional cardiovascular events without any increased risk of minor or major bleeding.39 Among patients with ACS without ST-elevation who did not undergo PCI, prasugrel was not found to be superior to clopidogrel in preventing ischaemic events.40
Ticagrelor
Unlike clopidogrel and prasugrel, ticagrelor is a cyclopentyl-triazolopyrimidine ADP antagonist and has distinct pharmacokinetic and pharmacodynamic properties. Ticagrelor binds directly to the P2Y12 receptor and alters its conformation, resulting in reversible inhibition. The drug does not require metabolic activation and thus exhibits a comparatively rapid onset and offset of effect, necessitating comparatively frequent dosing to achieve steady state ADP inhibition.41 Plasma levels of ticagrelor peak at 1.5–3.0 hours post-ingestion and reach steady state after 2–3 days.42 Although metabolic activation is not required for initiation of its antiplatelet effects, the drug does have an active metabolite, AR-C124910XX, which is produced following the interaction of the parent drug with cytochrome P450.41,42
Ticagrelor is administered as a loading dose of 180 mg, followed by maintenance dosing of either 60 or 90 mg twice daily. The Platelet Inhibition and Patient Outcomes (PLATO) study showed that ticagrelor was superior to clopidogrel in reducing mortality and further cardiovascular events in patients presenting with ACS, regardless of the presence or absence of CYP2C19 and CYP2C9 polymorphisms.43 The Dose Confirmation Study Assessing Anti-platelet Effects of AZD6140 versus Clopidogrel in Non–ST-Segment Elevation Myocardial Infarction-2 (DISPERSE-2) showed that there was no increase in major bleeding events in patients with non-ST segment ACS taking ticagrelor compared to clopidogrel; however, there were significantly more minor bleeding events.44 Ticagrelor achieves higher levels of platelet inhibition than clopidogrel,45 likely due to a combination of factors, including the aforementioned genetic variations in absorption and metabolism of clopidogrel.
In view of the greater platelet inhibition and consequent improved outcomes that are observed with prasugrel and ticagrelor compared with clopidogrel, many cardiology centres now recommend that the latter is not used as a first-line P2Y12 inhibitor in the management of acute STEMI.46
Cangrelor
Cangrelor is a novel P2Y12 inhibitor that, like ticagrelor, binds directly to the receptor and induces reversible blockade. The drug is a nonthienopyridine adenosine triphosphate analogue that is administered intravenously and has shown promising results in clinical trials to date.47 The drug has a rapid onset and offset of action, reaches steady state within a few minutes and achieves greater than 90 % inhibition of platelet activation resulting from the P2Y12 pathway.48
In a series of randomised controlled studies, when compared with current standard therapy, no significant differences in mortality or further MI were observed when patients were treated with either clopidogrel or cangrelor before49 or during PCI.50 A double-blind placebo-controlled trial involving 11,145 patients subsequently found that cangrelor significantly reduced the rate of ischaemic events during PCI, with no increase in severe bleeding, compared with clopidogrel.51 At present, cangrelor has been approved by US and European regulatory agencies for use in patients undergoing PCI, although it has not yet been recommended by the National Institute for Health and Care Excellence (NICE) for use in the UK due to a relative lack of clear data.52,53
Glycoprotein IIb/IIIa Inhibitors in Acute Coronary Syndrome
The platelet integrin complex glycoprotein (GP) IIb/IIIa represents the final common pathway of platelet activation.54,55 This molecule mediates platelet adhesion via binding to fibrinogen thereby forming bridges between platelets. A haemostatic platelet plug subsequently develops, which increases in size as further platelet activation is propagated.54 GP IIb/IIIa receptor inhibitors block this pathway and thus reduce thrombogenesis. Of the drugs in this class, abciximab (a monoclonal antibody fragment), tirofiban (a small, non-peptide molecule) and eptifibatide (a cyclic heptapeptide derived from rattlesnake venom) are used in clinical practice.55,56 Abciximab additionally binds to integrin receptors on leucocytes and endothelial cells, thus reducing the adhesion of platelets to these cells.56
GP IIb/IIIa inhibitors have played varying roles as antiplatelet agents over the past 20 years.55 Data from several large-scale meta-analyses looking at GP IIb/IIIa inhibitor clinical trials in the medical management of non-ST-elevation ACS indicate a significant reduction in further MI and overall mortality in patients treated with these agents,57,58 and although they were formerly used as key therapies in the management of acute MI for several years, GP IIb/IIIa inhibitors have been gradually phased out in favour of the P2Y12 inhibitors.
A similar trend has followed with regard to the use of GP IIb/IIIa inhibitors as prophylactic antithrombotic agents in patients undergoing PCI, where their use has been declining in favour of novel antithrombotic/ anticoagulant drugs. The TRITON-TIMI 38 study found that prasugrel significantly reduced the risk of cardiovascular events in patients with ACS after PCI regardless of whether or not a GP IIb/IIIa inhibitor was used concurrently.59 The Intracoronary Stenting and Antithrombotic Regimen–Rapid Early Action for Coronary Treatment (ISAR–REACT) trial enrolled 2,159 patients with coronary artery disease who underwent elective PCI following pre-treatment with clopidogrel 600 mg and either abciximab or placebo; there was no observable clinical benefit in those receiving abciximab over the 30 days post procedure.60 The ISAR–REACT 2 study subsequently assessed 2,022 patients with UA or NSTEMI undergoing PCI who were pre-treated with 600 mg clopidogrel and either abciximab or placebo, and found that abciximab significantly reduced the incidence of adverse events, although only in those patients with elevated troponin levels at presentation.61 In the UK, current NICE guidance recommends GP IIb/IIIa inhibitors as an adjunct to PCI for all patients with diabetes undergoing elective PCI, and for those patients undergoing complex procedures, but not for the routine management of ACS or PCI.62
Anti-inflammatory Effects of Antiplatelet Drugs: Possible Relevance to Acute Coronary Syndrome
In addition to their antiplatelet effects, it has become increasingly recognised that aspirin and the ADP receptor antagonists also exhibit beneficial anti-inflammatory effects in the context of ACS. The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) identified that elevated levels of high-sensitivity C-reactive protein (hsCRP), a hepatically synthesised biomarker of inflammation produced in response to macrophagederived interleukin-6, were associated with cardiovascular events including MI and stroke in patients with no known cardiac disease.63,64 HsCRP was found to be a stronger predictor of events than the traditionally used low-density lipoprotein levels, demonstrating the importance of underlying inflammation in the development of atherosclerosis.
Pro-inflammatory biomarkers have been shown to have significant prognostic value in STEMI,65 and a recent clinical trial has also identified a potential benefit of an anti-inflammatory therapy (colchicine) in the management of STEMI.66 Furthermore, levels of pro-inflammatory CD14highCD16+ monocytes are also associated with ACS, and a multitude of clinical studies have highlighted the association between raised levels of CD16+ monocytes and coronary disease. CD16+ monocyte counts are elevated in patients with UA compared with matched control subjects with stable coronary artery disease and, among the UA patients, those with intermediate–high risk of MI had significantly higher counts of the CD14highCD16 monocyte subset.67 Tapp et al. found a correlation between CD14highCD16+ counts and peak troponin-T levels post-STEMI, as well as a correlation between CD14highCD16+ counts and left ventricular ejection fraction post STEMI.68 A study of 951 patients referred for elective coronary angiography showed that a higher CD14highCD16+ count was predictive of cardiovascular events including MI, ischaemic stroke and death from cardiovascular causes.69 It has been demonstrated that both aspirin and clopidogrel exert immunomodulatory effects and are capable of counteracting the CD14highCD16+ monocyte increase that occurs in vivo under pro-inflammatory conditions,70 suggesting a potential role for these drugs in reducing monocytic biomarkers of inflammation in patients with cardiovascular disease and possibly thereby improving outcomes further in ACS.
Conclusion
Coronary artery disease and its related pathologies remain the leading cause of death worldwide. Antiplatelet agents have been used for decades to improve outcomes in patients with ACS and may do so not only through their antithrombotic properties but also through their anti-inflammatory effects, although their relative contribution in this context remains a subject of debate. What is clear, however, is that increased antithrombotic efficacy improves outcomes post-ACS, but carries the price of increased bleeding risk, so that at some point diminishing returns accrue from ever more efficacious antiplatelet therapy. The challenge for the future is to better predict the benefit:risk ratio in individual patients, so that the intensity of antiplatelet therapy can be optimised on a personalised basis.