







Pulmonary arterial hypertension (PAH) is a chronic and life-threatening disease characterised by progressive vascular remodelling that leads to increased pulmonary vascular resistance, right ventricular heart failure and death. PAH is defined by >25 mmHg increase in pulmonary arterial blood pressure and a pulmonary capillary wedge pressure of 15 mmHg.1 If left untreated PAH is fatal; it has a survival rate of just 34 % after 5 years.2 Current therapies for PAH include stimulating the nitric oxide (NO)–soluble guanylate cyclase (sGC)–cyclic guanosine monophosphate (cGMP) axis, improving the prostacyclin pathway or inhibiting the endothelin pathway.3,4
Although the causal relationship remains unproven, the NO–sGC–cGMP axis is ultimately a critical factor in the development of PAH because the condition is associated with endothelial dysfunction, impaired NO synthesis and insufficient stimulation of the NO–sGC–cGMP pathway.5 NO activates sGC, resulting in the synthesis of cGMP, which is a key mediator of pulmonary arterial vasodilatation that may also inhibit vascular smooth muscle proliferation and platelet aggregation. Dysregulation of the NO–sGC–cGMP axis results in pulmonary vascular inflammation, thrombosis and constriction, and ultimately leads to PAH.
Therapeutic options targeting the NO–sGC–cGMP axis include phosphodiesterase type 5 (PDE5) inhibitors, such as sildenafil and tadalafil, and the sGC stimulator riociguat.6 This review discusses the similarities and differences between PDE5 inhibitors and sGC stimulator and considers which is better for the treatment of PAH (Table 1).
Similarities in Pharmacological Action
Both classes of drug modify the cGMP-mediated signalling cascade. Increased levels of cGMP lead to vasodilatation and the inhibition of vascular smooth muscle proliferation and fibrosis. The presence of this nucleotide also exerts antithrombotic and anti-inflammatory effects. These effects are controlled by cGMP-dependent protein kinases, cGMP-dependent ion channels and phosphodiesterases.7,8 Activation of cGMP-dependent protein kinase isotype I (cGKI) decreases cytosolic Ca2+ concentration indirectly via the activation of membrane K+ channels, leading to the hyperpolarisation of vascular smooth muscle cell membrane. Additionally, cGKI phosphorylates vasodilator-stimulated phosphoprotein, an actin-binding protein whose phosphorylation status is related to the proliferation of vascular smooth muscle cells.9 These effects lead to the relaxation of smooth muscle cells.10 The cGKI also enhances canonical bone morphogenetic protein signalling via Smad1/5 and keeps pulmonary artery smooth muscle cells in a differentiated state with low proliferation. These effects are lost if there is a mutation in the bone morphogenetic protein type II receptor, as there is a reduction in downstream Smad1/5 phosphorylation.11,12
Increasing the amount of cGMP present through the use of PDE5 inhibitors or a sGC stimulator could lead to inhibition of the phosphodiesterase type 3 subtype, in turn leading to increased levels of cyclic adenosine monophosphate resulting in a positive inotropic effect.4 In addition to this effect, protein kinase G activation following the rise in cGMP level causes the mitochondrial KATP channels in cardiac cells to open,13,14 resulting in cardioprotective effects.15,16 These effects may improve right heart function in PAH.
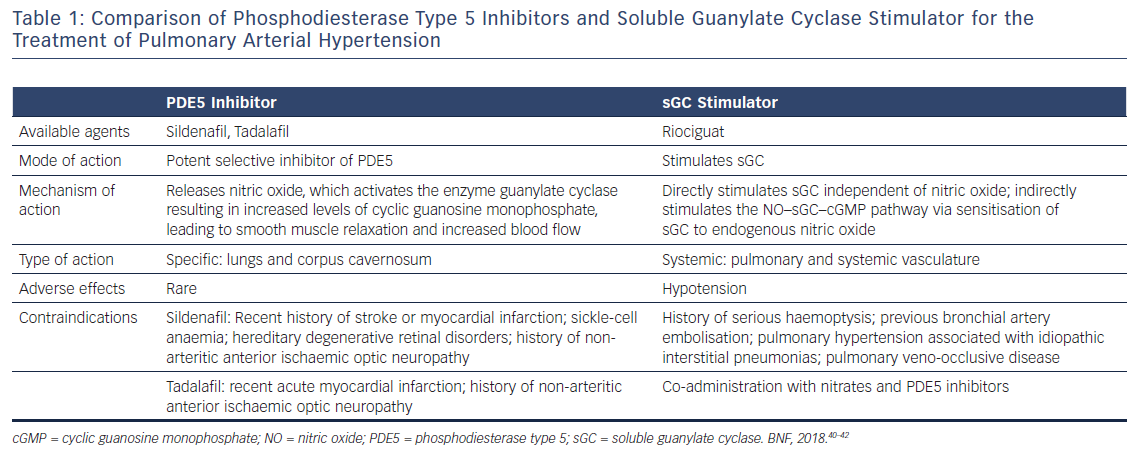
Differences in Pharmacological Action
PDE5 Inhibitors
The enzyme PDE5 is found in high concentrations in the pulmonary arteries and specifically degrades cGMP. It is important in the regulation of cGMP-specific signalling pathways, which effect smooth muscle contraction and relaxation, and is therefore a key target of PAH treatment. PDE5 inactivates cGMP by catabolising it to 5’GMP. In PAH, the expression and activity of PDE5 are enhanced in the smooth muscle cells of the pulmonary arteries.17–19 Enhancement of PDE5 activity and reduction in NO bioavailability both lower cGMP concentration, resulting in vasoconstriction, enhanced smooth muscle cell proliferation and the promotion of resistance to apoptosis.20
The PDE5 inhibitors sildenafil and tadalafil block the breakdown of cGMP. The resultant increase in cGMP concentration leads to relaxation of the smooth muscle and vasodilation. These effects are dependent on NO availability and sGC activity.21,22 PDE5 is distributed in the smooth muscles of the corpus cavernosum in penile tissue and in blood vessels in the lungs, which are the main sites of action of PDE5 inhibitors. The selectivity of these agents for lung tissue is maintained even when they are administered systemically.23,24
sGC Stimulator
The conversion of guanosine triphosphate to cGMP is catalysed by the enzyme sGC. This enzyme exists as a heterodimer, consisting of a larger α subunit and a smaller haem-binding subunit. In the resting state, the subunit contains a ferrous haem iron (Fe2+) that binds NO with picomolar affinity, enhancing sGC activity by several hundred-fold.25,26 Other circulating peptides, such as natriuretic peptide, can activate particulate guanylate cyclase to convert guanosine triphosphate to cGMP. The sGC stimulator riociguat has a dual mode of action, sensitising sGC to endogenous NO by stabilising NO–sGC binding and directly stimulating sGC via a different binding site. This drug hasbeen shown to produce cGMP independent of the presence of NO.27,28 As sGC is expressed in the pulmonary and systemic vasculature, the effect of riociguat is not limited to the pulmonary arteries and hypotension is often the dose-limiting adverse effect.
Supportive Evidence
PDE5 Inhibitors
Decreased endothelial NO production and increased PDE5 expression and activity are two important pathological features of PAH. Sildenafil and tadalafil are selective and potent inhibitors of PDE5 and increase intracellular cGMP levels. As PDE5 is expressed at high levels in the pulmonary circulation compared with systemic vessels, PDE5 inhibitors induce pulmonary vasodilation without decreasing systemic artery pressure. Sildenafil Use in Pulmonary Arterial Hypertension (SUPER), a double-blind, placebo-controlled, randomised study, demonstrated that sildenafil improved exercise capacity, World Health Organization functional class and haemodynamics in patients with symptomatic PAH.30 PDE5 inhibitors are widely used as an effective treatment for clinical PAH and their long-term safety and tolerability have been demonstrated by several randomised clinical trials.31 It is also strongly suggested that earlier treatment with sildenafil could bring better outcome in the treatment for PAH.31 Tadalafil has a longer half-life than sildenafil;32 however, a recent analysis designed to estimate the costs and quality-adjusted life years associated with bosentan, ambrisentan, riociguat, tadalafil, sildenafil and supportive care for PAH in treatment-naive patients suggested that sildenafil was the most cost-effective therapy in patients with functional class II or III PAH.33 Despite this, tadalafil was found to be less costly and more effective than supportive care in these patients.
sGC Stimulator
Endothelial NO synthase expression, NO production and NO availability are substantially reduced in patients with PAH,5,10 so the sGC stimulator may be effective in patients who have not sufficiently responded to a PDE5 inhibitor.34–36 The recent open-label, uncontrolled, phase IIIb Riociguat Clinical Effects Studied in Patients with Insufficient Treatment Response to PDE5 Inhibitor (RESPITE) study suggested that switching from PDE5 inhibitors to riociguat improved a range of clinical and haemodynamic endpoints in patients with PAH who have had an inadequate response to PDE5 inhibition.34 Patients with PAH-systemic sclerosis (SSc) tend to have a worse prognosis than patients with idiopathic PAH, and have poorer responses to treatment and worse outcomes than those whose PAH is associated with other connective tissue diseases (CTDs). However, the long-term extension of the Pulmonary Arterial hyperTENsion sGC-stimulator Trial (PATENT-2) demonstrated the same survival at 2 years (93 %) for those with PAH-CTD and those with idiopathic or familial PAH who were receiving treatment with riociguat, despite more than half of the PAH-CTD patients having PAH-SSc.37 Since SSc is characterised by systemic vascular disorder, the action of a non-selective vasodilator such as a sGC stimulator may be more favourable than a pulmonary artery-selective vasodilator. A randomised, double blind, placebo-controlled phase II study to investigate the efficacy and safety of riociguat in patients with diffuse cutaneous SSc is underway.38
Combination Treatment
Upstream NO–sGC activity and downstream PDE5 activity affect the level of cGMP. Even if the degradation of cGMP is completely blocked by a PDE5 inhibitor, there will not be sufficient cGMP produced if there is insufficient NO produced upstream. In the same way, even if sGC stimulator enhances the production of cGMP, the cGMP level cannot be maintained if its degradation has been enhanced by excessive PDE5 activity. The combination of sGC stimulator and a PDE5 inhibitor would therefore appear to be an attractive therapeutic option. However, combination therapy with riociguat and sildenafil had no favourable effects on exploratory clinical parameters, including haemodynamics and exercise capacity, in patients with PAH in the PATENT PLUS study.39 There were high rates of discontinuation due to hypotension and three deaths – one due to cardiac arrest, one from right heart failure and one from cerebral bleeding due to a fall – occurred during the study. The possibility that the combination of medications contributed to the fall could not be excluded. There were potentially unfavourable safety signs with sildenafil plus riociguat and no evidence of a positive benefit–risk ratio. Therefore, concomitant use of riociguat with PDE5 inhibitors is contraindicated.
Conclusion
Based on recent cost-utility analysis, sildenafil may be recommended as the first choice and tadalafil as second choice in patients with functional class II and III PAH. However, some patients may gain more benefit taking tadalafil, with its once-daily administration, rather than sildenafil, which needs to be taken a three times per day. Patients with insufficient response to PDE5 inhibitors should be switched to the sGC stimulator riociguat. Patients with PAH-SSc may gain further benefit from riociguat due to its systemic vascular action.
While there are clear pharmacokinetic and pharmacodynamic differences between these agents, it is still difficult to determine which agent is most appropriate for a specific PAH patient. Some patients respond better to sGC stimulator than a PDE5 inhibitor and vice versa. Additional data from clinical trials are needed to clarify which treatment is best; a randomised controlled trial to further investigate switching from PDE5 inhibitors to riociguat is underway (REPLACE: NCT02891850).