Chronic heart failure is the leading cause of hospitalisation in Germany1 and other European countries. It is the result of various cardiovascular diseases, such as myocardial infarction, arterial hypertension and valvular heart diseases. About 50 % of patients with heart failure have normal systolic, but impaired diastolic function, a condition termed heart failure with preserved ejection fraction (HFpEF), in contrast to heart failure with reduced ejection fraction (HFrEF).2,3 Patients with ischaemic heart disease and/or myocardial infarction are more likely to develop HFrEF, while HFpEF more likely inflicts the elderly, female gender and patients with arterial hypertension.4 While initially it seemed that patients with HFpEF only had a slightly better prognosis than patients with HFrEF (by 4 %),3 more recent evidence suggests that prognosis of HFpEF per se is clearly less adverse than of HFrEF (by 32 %), while frequent co-morbidities affect overall prognosis of patients with HFpEF.4,5 Over the past decades, advancements in the treatment of patients with HFrEF led to continuous improvements of overall prognosis, while in HFpEF, most drugs used in patients with HFrEF were not effective.3,6 In the 2012 European Society of Cardiology (ESC) Guidelines for the diagnosis and treatment of acute and chronic heart failure, definite recommendations for the pharmacological treatment of patients with HFrEF are provided, while recommendations for patients with HFpEF are scarce.6 Hence, we herein will focus on the pharmacological treatment of patients with HFrEF and for simplicity will refer to them as patients with ‘heart failure’ (HF).
Pathophysiology of Heart Failure
Blood pressure (BP) is defined as the product of cardiac output (C.O.) and systemic vascular resistance (SVR), where C.O. is the product of stroke volume (SV) and heart rate (HR). Systolic dysfunction of the left ventricle (LV) reduces SV and thus C.O., which results in decreased BP. A decrease in BP is sensed by baroreceptors in the carotid artery and the aorta, which activates the sympathetic nervous system that is centrally controlled in the brain stem (medulla oblongata), triggering increased release of norepinephrine in the myocardium and of epinephrine from the adrenal glands to the bloodstream (see Figure 1). While epinephrine increases SVR via vascular alpha (α)-adrenergic receptors, norepinephrine has positive inotropic and chronotropic effects in the heart by stimulating beta (β)-adrenergic receptors, increasing HR and SV. Together, sympathetic activation increases BP and therefore has beneficial effects on short-term haemodynamics while in the long run, leads to LV remodeling and dysfunction7 through activating pro-hypertrophic signaling pathways8 and inducing apoptosis.9,10 Furthermore, elevated heart rate per se is associated with adverse prognosis in patients with HF.11
Activation of renal sympathetic efferent nerves to the kidney with subsequent activation of β-adrenergic receptors as well as reduced renal blood flow increase the release of renin from the kidney, which converts angiotensinogen to angiotensin I, which is then converted to angiotensin II (Ang II) by the angiotensin-converting enzyme (ACE;see Figure 1). Ang II induces vasoconstriction via type 1 Ang II (AT1) receptors and stimulates the release of aldosterone from the adrenal glands. Aldosterone, in turn, increases sodium (Na+) and water (H2O) retention in the kidney, which elevates intravascular volume and hence, SV and BP. Similar to the sympathetic nervous system, the activation of the renin-angiotensin-aldosterone system (RAAS) has beneficial short-term effects on BP, but adverse long-term effects on LV remodeling and prognosis, which is largely related to the activation of pro-hypertrophic and maladaptive signaling pathways in the heart.12
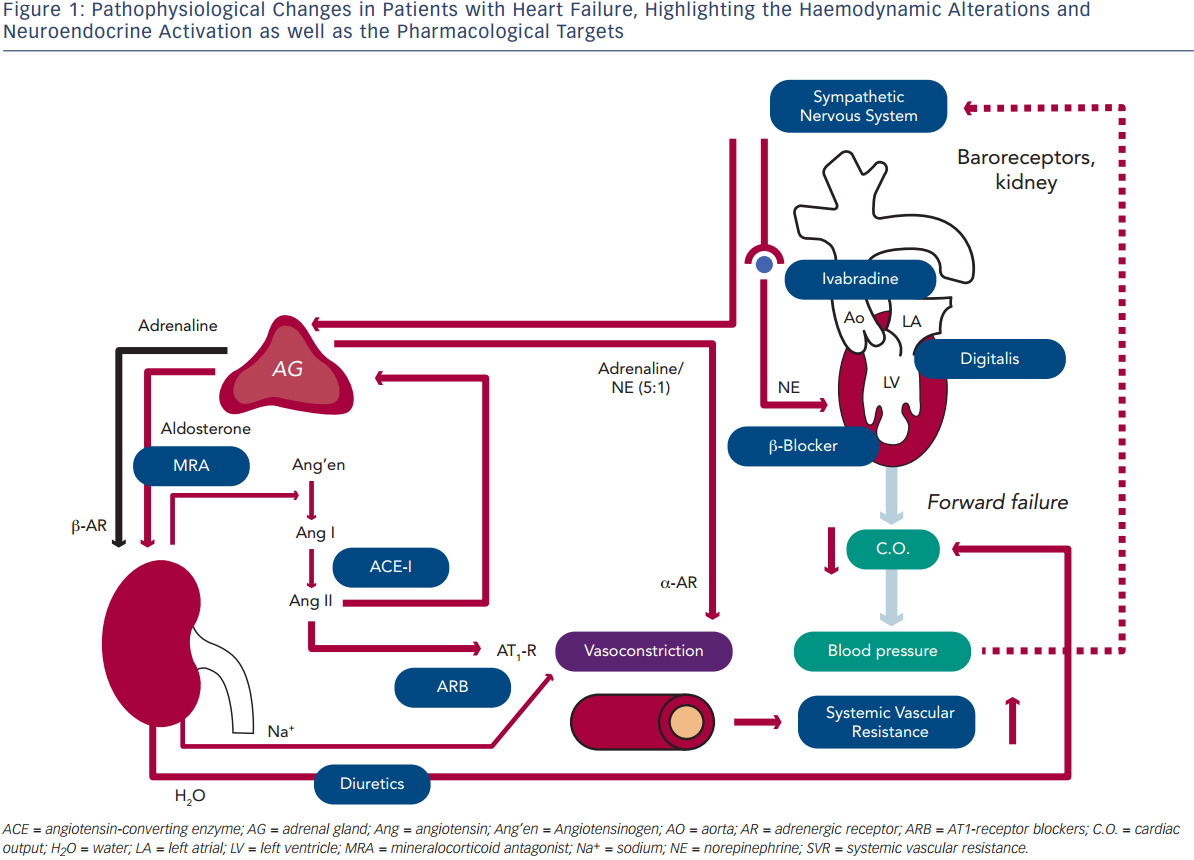
With progressive LV remodeling and dysfunction, the filling pressure of the LV increases, which triggers the production and release of brain natriuretic peptide (BNP) from LV myocardium, with the N-terminal pro-BNP being used as the currently most sensitive and reliable biomarker for the haemodynamic status and prognosis of patients with HF. Through its natriuretic action, BNP tends to antagonise the effects of RAAS activation. As a consequence of elevated LV filling pressures, left atrial pressure increases, inducing atrial dilation and fibrosis and thus, providing a substrate for atrial fibrillation (AF).13 Furthermore, elevated LV and atrial filling pressures trigger pulmonary congestion, which is the basis for the leading symptom of HF (i.e. dyspnoea). When pulmonary congestion continues to increase pulmonary artery pressures, the right heart is also exposed to elevated afterload and thus, after a prolonged time, right ventricular (RV) decompensation may lead to the development of oedema in arms, legs and potentially the gut.
Considering these pathophysiological changes during HF, the treatment of these patients primarily aims to antagonise neuroendocrine activation and congestion (see Figure 1), which in concert trigger maladaptive remodeling of the LV.12
Pharmacological Treatment
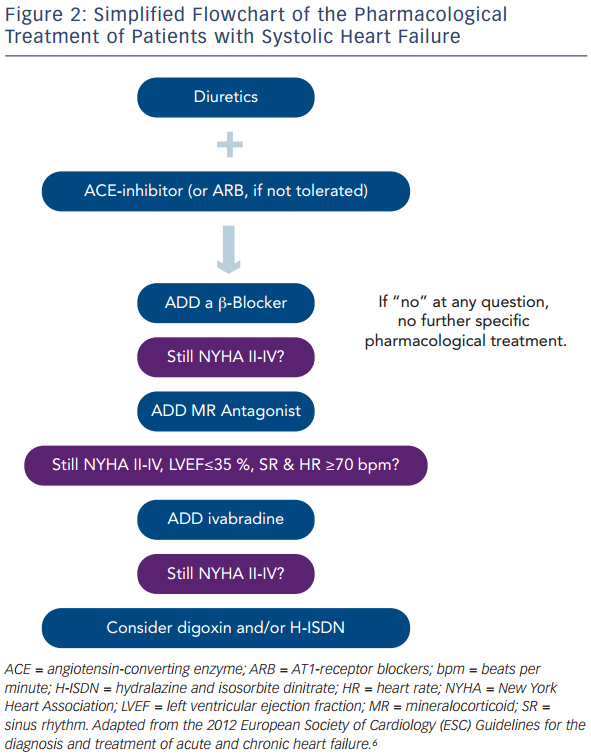
The major aims of treating patients with HF are to relieve symptoms, prevent hospitalisation, and to improve functional capacity, quality of life and survival.6 Drugs that fulfil these aims frequently also ameliorate LV remodeling and lower circulating natriuretic peptides. The cornerstones of medical treatment are diuretics, ACE-inhibitors or AT1-receptor blockers (ARB), β-adrenergic receptor antagonists (β-blockers) and mineralocorticoid antagonists (MRA; see Figure 1). Furthermore, in some patients with sinus rhythm in whom HR reduction is insufficient despite β-blockade, the use of ivabradine is justified, and digitalis glycosides may be useful to improve morbidity and to control HR in patients with AF. In the following paragraphs, we give an overview on the use of these drugs, their mechanisms of action, and in particular, the clinical evidence supporting their use.
Diuretics
Although the effects of diuretics on morbidity and mortality were never tested in patients with HF, they are absolutely essential for decongestion and thus, improvement of symptoms. Loop diuretics are more effective in inducing diuresis compared with thiazides and can be used intravenously during acute decompensation or orally during stable phases of disease to maintain euvolaemia (patient´s ‘dry weight’). The most commonly used diuretics are furosemide and torasemide, whose doses can be (self-) adjusted depending on signs of fluid retention (monitored by body weight).6 Since ACE-inhibitors/AT1-antagonists and MRAs, and in particular the combination of these, can elevate serum potassium, loop diuretics or thiazides are better suited than potassium-sparing diuretics in patients with HF.6
Angiotensin-converting Enzyme Inhibitors
The two major trials that established the use of ACE-inhibitors in the treatment of HF are the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS)14 and the Studies of Left Ventricular Dysfunction (SOLVD)15 trials. In CONSENSUS, enalapril was tested against placebo in 253 patients with HF in New York Heart Association (NYHA) functional class IV on a stable background therapy consisting of vasodilators and diuretics for a follow-up period of 188 days. Enalapril improved survival by 31 % and also symptoms, but had no effect on sudden cardiac death.14 The fact that enalapril improved prognosis in patients with or without concomitant use of other vasodilators indicated that the beneficial effect of enalapril was the result of reducing Ang II levels (which may ameliorate LV remodeling) rather than of pure haemodynamic improvement (reduction of afterload).14 Since CONSENSUS was performed in severely ill patients (NYHA IV),14 in the SOLVD study enalapril was compared with placebo in 2,569 patients with less severe HF (NYHA functional class II-III) and left ventricular ejection fraction (LVEF) ≤35 %, on a background therapy consisting of spironolactone and digitalis.15 Enalapril reduced total mortality by 16 %, aggravation of HF by 22 % and death or hospitalisation for HF by 26 %, respectively.15 Again, no benefit on sudden cardiac death was achieved.15 It is important to up-titrate ACE-inhibitors to the maximal tolerable dose, since a trial with lisinopril (Assessment of Treatment with Lisinopril and Survival [ATLAS]) indicated a benefit of high-dose over low-dose lisinopril in NYHA functional class II-III HF patients.16 Further support for the use of ACE-inhibitors in HF comes from a metaanalysis17 and three larger trials in patients with HF, LV dysfunction or both after myocardial infarction (Survival and Ventricular Enlargement trial [SAVE], Acute Infarction Ramipril Efficacy [AIRE] and Trandolapril in Cardiac Evaluation [TRACE]).18 Finally, ACE-inhibitors are the only drugs with a proven benefit in asymptomatic patients with HF based on a 20 % relative risk reduction (RRR) in the SOLVD Prevention trial.19 ACE-inhibitors should only be used in patients with sufficient renal function (i.e. creatinine ≤2.5 mg/dl or estimated glomerular filtration rate [eGFR] ≥30 mL/min/1.73 m2 and normal potassium levels).6
Beta-blockers
A landmark study about the use of β-blockers in patients with HF was published by Waagstein et al.20 in 1975, who reported improved symptoms and LV function in seven patients with dilated cardiomyopathy in response to β-blockers. At that time, β-blockers were strictly contraindicated in patients with HF since it was assumed that the relief of sympathetic activation would deprive the heart of a critical stimulus to maintain contractility. In fact, acute administration of β-blockers in patients with HF can lead to a transient deterioration of C.O.,21 while a more long-term treatment typically increases LVEF21 and reverses remodeling of the LV (decreasing LV chamber size).22 On a cellular level, improvements of cardiac function by β-blockers are associated with a restoration of cardiomyocyte calcium handling proteins and contractile filaments.23 After promising (but not yet significant) results with the second-generation β-blockers, metoprolol tartrate and bisoprolol in the Metoprolol in Dilated (MDC)24 and Cardiac Insufficiency Bisoprolol Study (CIBIS) trials25 in the early 1990s, a study programme with the third-generation β-blocker carvedilol was terminated early due to a 65 % reduction in overall mortality by carvedilol versus placebo.26 In subsequent years, larger and better designed randomised clinical trials confirmed the benefits of β-blockade with metoprolol succinate (Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure [MERIT-HF]),27 bisoprolol (CIBIS II)28 and carvedilol (Carvedilol Prospective Randomized Cumulative Survival [COPERNICUS]).29 Importantly, the benefits of β-blockers (~34 % RRR of all-cause mortality by metoprolol succinate,27 bisoprolol28 and carvedilol,29 respectively) were generated on the background of ACE-inhibitor therapy (>90 % of patients) and were overall comparable among the three mentioned agents. Although one trial reported superiority of carvedilol over metoprolol (Carvedilol or Metoprolol European Trial [COMET]30), it has to be taken into account that this trial30 was conducted with short-acting metoprolol tartrate, while the successful MERIT-HF trial27 was conducted with long-acting metoprolol succinate. Further support for the benefit of β-blockers comes from the Study of Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial,31 in which the third-generation β-blocker nebivolol was tested versus placebo in elderly (≥70 years) patients with HF, of whom 36 % had a LVEF of >35 %. Nebivolol reduced the combined endpoint of death or cardiovascular mortality, but did not reduce mortality.31 Bucindolol, another third-generation β-blocker with partial agonist activity32 did not reduce mortality in the Beta-Blocker Evaluation in Survival Trial (BEST).33
Taken together, the 2012 ESC Guidelines on the treatment of HF recommend the use of bisoprolol, carvedilol, metoprolol succinate or nebivolol in patients with HF (Class I, Level A recommendation). It is important to start on a low dose (to prevent initial deterioration of HF) and up-titrate the drug to the maximally tolerated dose, ideally aiming at a HR between 60 and 70 beats per minute (bpm). During decompensation of a patient with HF, continuation of the β-blocker is safe, although a dose reduction may be required.34
Mineralocorticoid or Aldosterone Receptor Antagonists
Currently there are two lead compounds of this class on the market, spironolactone and eplerenone. While the former is less specific and antagonises mineralocorticoid receptors, the latter is more specific for aldosterone receptors. Together, they are classified as MRAs. The first randomised placebo-controlled outcome study with a MRA was the Randomized Aldactone Evaluation Study (RALES),35 in which HF patients in NYHA functional class III and an LVEF ≤35 % were randomised to spironolactone (25–50 mg once daily) or placebo added to conventional treatment, at that time consisting predominantly of diuretics (100 %), ACE-inhibitors (95 %) and digitalis (~75 %). Compared with placebo, spironolactone improved symptoms, decreased hospitalisation for worsening HF (by 35 %) and overall mortality (by 30 %).35 Of note, this improved survival was due to both reduction of death from progressive HF and sudden death from cardiac causes.35 While hyperkalaemia was rare, gynaecomastia as a side effect occurred in 10 % versus 1 % of patients treated with the MRA versus placebo, respectively.35
In the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS),36 treatment of patients with acute myocardial infarction complicated by LV dysfunction (LVEF ≤40 %) and HF with the more specific aldosterone antagonist eplerenone reduced overall and cardiovascular mortality by 15 % and 17 %, respectively. Furthermore, hospitalisations for cardiovascular or any reasons were reduced. Interestingly, eplerenone also reduced sudden cardiac death by 21 %.36 An advantage of EPHESUS36 over RALES35 was that in the former, a much higher percentage of patients were treated with β-blockers (75 % versus 11 %).
To finally resolve whether, also in patients with stable HF, MRAs would improve prognosis added to a background therapy that included β-blockers, and whether MRAs were efficient also in patients with mild symptoms of HF (RALES was performed in NYHA functional class III patients), the recent Eplerenone in Mild Patients Hospitalization And Survival Study in Heart Failure study (EMPHASIS-HF)37 enrolled 2,737 patients in NYHA functional class II and a LVEF ≤35 %. In fact, eplerenone reduced the primary composite endpoint of death from cardiovascular causes or hospitalisation for HF by 37 %, and cardiovascular as well as overall mortality per se by 24 %, respectively. Hospitalisations for heart failure and for any cause were also reduced with eplerenone. Mild hyperkalaemia (serum potassium [K+] >5.5 mmol/L) was (as expected) somewhat more frequent (+64 %) in eplerenone- compared to placebo-treated patients, while hypokalaemia was less frequent with eplerenone.37
Taken together, MRAs reduce morbidity and mortality in patients with HF, but also acute myocardial infarction complicated by LV dysfunction and HF. Both MRAs can provoke hyperkalaemia and worsen renal function, while spironolactone, but not eplerenone, can induce gynaecomastia as a side effect. Thus, in the current ESC guidelines, MRAs are recommended in addition to ACE-inhibitors and β-blockers in all patients with HF with a LVEF ≤35 % and maintaining symptoms (NYHA II-IV).6 However, similar to ACE-inhibitors, MRAs should be limited to patients with adequate renal function and normal serum K+ concentrations.6
AT1-Receptor Antagonists
A common side effect of ACE-inhibitors is to provoke dry cough due to inhibition of bradykinin breakdown by chymase. Furthermore, some Ang II can still be generated despite ACE-inhibition through the so-called ‘escape phenomenon’. The rationale behind the development of ARBs was to circumvent these limitations of ACE-inhibitors. Although ARBs produced less side effects than ACE-inhibitors (in particular, cough), they were not superior to ACE-inhibitors in terms of survival in the Losartan Heart Failure Survival Study (ELITE II).38 However, when patients with HF did not tolerate treatment with an ACE-inhibitor, treatment with ARBs reduced morbidity and mortality when compared with placebo, as revealed by the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) Alternative trial39 and a subgroup analysis40 of the Valsartan Heart Failure Trial (Val-HeFT).41 In this scenario (HF patients intolerant to ACE-inhibitors), and in analogy to ACE-inhibitors in the ATLAS trial,16 the Heart Failure Endpoint Evaluation with the Angiotensin II Antagonist Losartan (HEAAL) trial42 revealed that a higher dose of losartan (150 mg) was superior to a lower dose of the same drug (50 mg) in preventing HF hospitalisation. In contrast, the addition of ARBs on top of ACE-inhibitors did not improve overall mortality in the Val-HeFT41 or the CHARM-Added43 trials. However, cardiovascular mortality was improved by 16 % with candesartan in CHARM-Added trial.43 Furthermore, both trials showed reduced hospitalisation for HF deterioration (by 24 % in Val-HeFT and 17 % in CHARM-Added), whereas all-cause hospitalisation remained unchanged. While 35 % (in Val-HeFT) and 55 % of patients (in CHARM-Added) had a β-blocker in their background medication, only a few patients were treated with a MRA.41,43 Importantly, a post hoc analysis of Val-HeFT revealed that the ARB was particularly efficient in patients not taking a β-blocker or ACE-inhibitor (or neither), whereas it was adverse in those patients taking both.41 Since in the EMPHASIS trial with eplerenone, the MRA efficiently reduced mortality and morbidity when added to ACE-inhibitors and β-blockers,37 while ARBs did not achieve this,41,43 ARBs no longer remain a first-choice recommendation in patients with HF, while they received a class I level A recommendation in the current guidelines as an alternative in patients intolerant to an ACE-inhibitor.6
Ivabradine
In patients with HF, elevated HR is associated with an adverse prognosis. For instance, HF patients with HR ≥87 bpm have a more than two-fold worse prognosis than patients with a HR between 70 and 72 bpm.11 Since β-blockers, besides their negative chronotropic effects, also have negative inotropic effects8 that may acutely deteriorate C.O.,21 ivabradine was developed as a selective HR-lowering drug by inhibiting the current of the ‘funny’ channel (If), which triggers slow depolarization in sinus node cells. Accordingly, ivabradine reduces HR only in patients with sinus rhythm (SR), while in patients with AF, it has no effect. The prevailing pathophysiological view has long been that HR reduction is beneficial for the heart primarily due to lowering oxygen consumption. However, more recent experimental and clinical evidence also indicates that HR reduction improves vascular function and through this, unloads the heart. In animal models of atherosclerosis, HR reduction prevented vascular oxidative stress and endothelial dysfunction, reduced atherosclerotic plaque formation and stimulated collateral artery growth through improving the bioavailability of nitric oxide and reducing inflammation.44,45 Oxidative stress and inflammation are of particular importance in diastolic HF (HFpEF), where coronary microvascular inflammation is thought to induce hypertrophy and myofilament stiffness.46 Accordingly, HR reduction with ivabradine improved vascular stiffness and LV systolic and diastolic function in a mouse model of HFpEF.47 However, also in patients with systolic HF, ivabradine reduced arterial stiffness, which increased stroke volume by unloading the LV.48,49Taken together, these data indicate that ivabradine may improve myocardial pathologies through actions on the heart and the vessels.
The first large randomised placebo-controlled trial with ivabradine was performed in patients with coronary artery disease and a LVEF <40 % (Morbidity-mortality evaluation of the If inhibitor ivabradine in patients with coronary artery disease and left ventricular dysfunction [BEAUTIFUL] trial50). In this trial, 87 % of patients were receiving a β-blocker, and mean HR at baseline was only 72 bpm and hence, the average (placebo-corrected) HR reduction by ivabradine was only six bpm. Accordingly, the primary endpoint (a composite of cardiovascular death, admission to hospital for acute myocardial infarction and admission to hospital for new onset or worsening heart) was completely unaffected by ivabradine (hazard ratio 1.0).50 However, in a pre-specified subgroup analysis of patients with a HR of ≥70 bpm at baseline, ivabradine reduced secondary endpoints, such as admission to hospital for fatal and non-fatal myocardial infarction (by 36 %) and coronary revascularisation (by 30 %).50
Based on these positive, but rather hypothesis-generating data,50 the Systolic Heart failure treatment with the If inhibitor ivabradine trial (SHIFT)51 assigned patients with stable HF (NYHA functional classes II-IV, LVEF≤35 %) who were in SR with a HR of ≥70 bpm to ivabradine or placebo, respectively. Background medication included ACE-inhibitors or ARBs (together 93 %) and MRAs (60 %). While 90 % of patients were treated with a β-blocker, only 26 % of these patients received the full recommended dose. Ivabradine reduced the composite endpoint of cardiovascular death or HF hospitalisation by 18 %, which was driven primarily by a 26 % reduction of HF hospitalisation, while reductions of cardiovascular or all-cause mortality were not significant.51 The most common side effects of ivabradine versus placebo were bradycardia (5 % versus 1 %) and visual side effects (so-called phosphenes; 3 % versus 1 %).
Digoxin and Other Digitalis Glycosides
A central deficit in patients with HF is that in cardiac myocytes, calcium ions (Ca2+) handling is impaired, which is a causal factor for contractile dysfunction.52 A classical treatment of patients with HF is the use of digitalis glycosides, which inhibit the Na+/K+ adenosine triphosphatase (ATPase) and thus, elevate intracellular Na+ concentrations. This, in turn, increases Ca2+ influx via the Na+/Ca2+ exchanger, which increases cardiac contractility. As a second effect, digitalis glycosides prolong the refractoriness of the atrioventricular (AV)-node, which accounts for negative chronotropic effects in patients with AF, but less in SR (contrary to ivabradine). The only large randomised placebo-controlled trial with a glycoside was the Digitalis Investigation Group (DIG) trial,53 in which digoxin was compared with placebo in 6,800 patients in NYHA functional classes II-IV and a LVEF ≤45 %, added to a diuretic and an ACE-inhibitor. Published in 1997, this trial was before the time when β-blockers were widely used in patients with HF. The main outcomes of this trial were that digoxin reduced hospital admission for HF by 28 %, but did not affect all-cause mortality over three years.53
Combination of Hydralazine and Isosorbite Dinitrate
A characteristic hallmark in patients with HF is that through neuroendocrine activation, SVR is elevated, imposing an increased afterload to the failing heart. Thus, a therapeutic approach (that proved beneficial with ACE-inhibitors) is to lower SVR by applying vasodilating agents. Three trials investigated the effects of the vasodilatory combination of hydralazine and isosorbite dinitrate (H-ISDN) in patients with HF: first Vasodilator Heart Failure Trial (V-HeFT I),54 second Vasodilator-Heart Failure Trial (V-HeFT II)55 and African-American Heart Failure Trial (A-HeFT).56 In the Valsartan Heart Failure Trial (Val-HeFT I),54 H-ISDN increased exercise capacity and LVEF compared with placebo and showed a trend towards reduction in all-cause mortality.54 However, in that trial, no patient was treated with an ACE-inhibitor or a β-blocker. In V-HeFT II,55 H-ISDN was compared with the ACE-inhibitor enalapril, being inferior by tending to increase mortality by 28 % versus the ACE-inhibitor. A-HeFT compared H-ISDN to placebo added to a background medication including ACE-inhibitors or ARB (combined 84 %), digoxin (60 %), β-blockers (74 %) and spironolactone (39 %) exclusively in African- American patients with HF (NYHA III-IV).56 H-ISDN reduced mortality by 43 % and HF hospitalisation by 33 % and improved quality of life.56 Since results in white patients are currently inconclusive, the use of H-ISDN is recommended only for black patients and thus, uncommon in most European countries, while it is more common in the US.
Drugs Not Recommended
Despite its accepted benefit in patients with coronary artery disease, statins have not improved outcome in patients with HF. Rosuvastatin was neither efficient in patients with ischaemic nor non-ischaemic cardiomyopathy, based on the data of the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA)57 and the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico- Heart Failure (GISSI-HF) trial.58 Also, the renin inhibitor aliskiren was not efficient in decreasing rehospitalisation or cardiovascular death in patients with systolic HF.59 Drugs that could produce harm in patients with HF include thiazolidinediones (glitazones), calciumchannel blockers, non-steroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) inhibitors and therefore should be avoided.6 Finally, the addition of an ARB to an ACE-inhibitor and a MRA is not recommended due to the risk of renal dysfunction and hyperkalaemia.6
Summary
Taken together, patients with HF should be treated with diuretics to prevent or treat symptoms of congestion, and with an ACE-inhibitor, β-blocker and MRA to improve morbidity and mortality. For some patients, the use of ivabradine or digoxin and/or H-ISDN may be useful. In Figure 2, a simplified scheme adapted from the current guidelines on the treatment of patients with chronic systolic HF is illustrated that helps to choose the right drugs at the right time in the treatment of these patients.