










Until the 20th century, conventional medicine relied almost exclusively on observable characteristics to classify and treat human disease.1 Even in the modern era, physicians define diseases using phenotypic similarities and employ relatively simple algorithms to interpret diagnostic tools and plan treatment. The advent of low-cost genetic sequencing and its introduction into clinical care provides new insights into pathogenesis and improves the recognition of inherited diseases and complex associated entities. In this article, we review a relatively new concept, arrhythmogenic cardiomyopathy (AC), and explain why it is an exemplar for a more nuanced approach to diagnosis that promises more effective treatments.
The Paradigm of Arrhythmogenic Right Ventricular Cardiomyopathy
In 1982, Marcus et al. described a group of patients with right ventricular dysplasia who presented with ventricular tachycardias of left bundle branch block configuration, T-wave inversion in the right precordial leads and structural abnormalities of the right ventricle (RV).2 In cases of sudden death, histological examination showed fibrofatty replacement of the right ventricular myocardium combined with signs of myocardial degeneration and necrosis, with and without inflammation, which led to the adoption of the term ‘arrhythmogenic right ventricular cardiomyopathy’ (ARVC), rather than ‘dysplasia’ to describe this new potentially lethal myocardial disorder.3
The familial nature of the disease was recognised very early and inspired the identification of the first genetic mutation in ARVC, which was a two-nucleotide pair deletion in the junction plakoglobin (JUP) gene in patients with Naxos disease, a recessive syndromic form of ARVC associated with palmoplantar keratoderma and woolly hair.4,5 Around the same time, a recessive mutation in the desmoplakin (DSP) gene was identified as the cause of an autosomal recessive cardiocutaneous syndrome in Ecuadorian families known as Carvajal syndrome.6–8 Both JUP and DSP are key components of the myocyte intercalated disc, and candidate gene evaluation in families with more common dominant forms of ARVC soon revealed mutations in DSP and other desmosomal genes, including plakophilin-2 (PKP-2), desmocollin-2 (DSC-2) and desmoglein-2 (DSG-2), forming the concept of ARVC as a disease of the desmosome.9
The Emergence of Nosological Confusion
Even in early reports of ARVC, fibrous/fibrofatty lesions were found in the left ventricular myocardium, as well as the right.2,3 Electrocardiograms from patients with ARVC often demonstrated features consistent with left ventricular involvement and cardiac imaging occasionally demonstrated coexistent wall motion abnormalities, dilation and systolic dysfunction.2,5 With the widespread use of cardiac MRI to assess patients with suspected ARVC, it became apparent that subtle left ventricle (LV) disease in the form of myocardial fibrosis is almost the norm, particularly in patients with mutations in DSP and DSG2.10 In response to this, many investigators started to use terms such as ‘arrhythmogenic left ventricular cardiomyopathy’, and even more confusingly, ‘left-dominant arrhythmogenic cardiomyopathy’, to describe this broader spectrum of disease. However, this effort to preserve the concept of ARVC as originally described in the 1980s and 1990s has become increasingly tenuous with the demonstration of overlapping phenotypes, such as dilated cardiomyopathy and non-desmosomal gene mutations, such as phospholamban (PLN), transmembrane protein 43 (TMEM43), filamin C (FLNC), desmin, cadherin 2 (CDH2), lamin A/C (LMNA) and titin (TTN) in individuals fulfilling current diagnostic criteria for ARVC, as well as other phenotypes (Figure 1).11–17
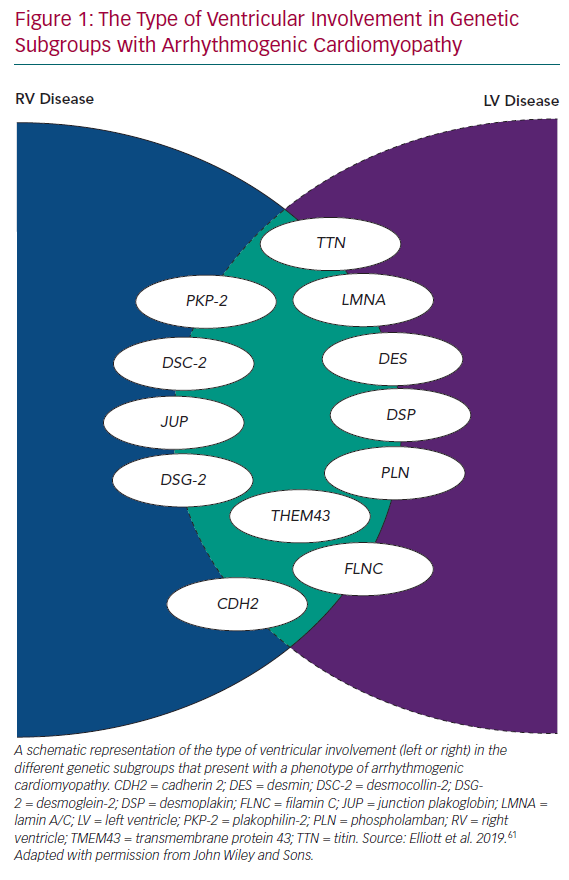
The Arrhythmogenic Cardiomyopathy Phenotype
Phenotypes describe a set of identifiable clinical characteristics that help to distinguish affected individuals from unaffected. The term AC represents an evolution of diagnostic terms describing disease of the LV and RV, but it was only recently that the term was formally used to describe this disorder.18
Although diagnostic criteria for the expanded AC spectrum do not yet exist, it is possible to summarise the key components of the AC phenotype:
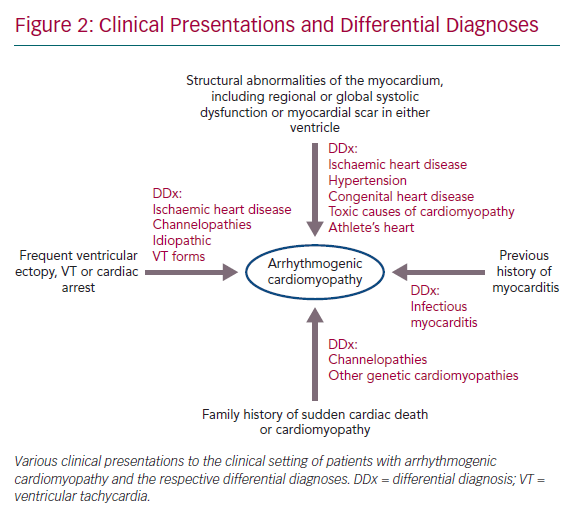
- Structural abnormalities of the myocardium, including regional or global systolic dysfunction or myocardial scar in either ventricle, unexplained by ischaemic heart disease, hypertension or an acute inflammatory trigger, such as viral infection or exposure to a toxic agent.
- Frequent ventricular ectopy, sustained or non-sustained ventricular tachycardia (VT) or unexplained cardiac arrest.
- A family history of sudden cardiac death or cardiomyopathy and a history of myocarditis presentation are also key features (Figure 2).
When defined in this way, the term AC serves as the first step towards disease classification and elucidation of aetiology. Contemporary studies show that AC encompasses a broad spectrum of genetic and non-genetic diseases, each with its own characteristic features and clinical course. A detailed description of all contenders for the label of AC is beyond the scope of this article, but two groups deserve emphasis due to their potential to cause premature death (often in young people) and their influence on therapy.
Arrhythmogenic Dilated Cardiomyopathy
It is important to appreciate that, by definition, the AC phenotype overlaps with that of dilated cardiomyopathy (DCM). DCM is currently defined only by LV size and function. However, a subgroup of patients characterised by syncope, rapid non-sustained ventricular tachycardia, increased ventricular ectopy and a family history of sudden cardiac death has been described.19 The term ‘arrhythmogenic DCM’ has been used to describe these cases, but in practice it aligns with the definition of AC.20
Arrhythmogenic DCM includes many genetic forms that are characterised mainly by dilatation and dysfunction of the LV. Examples include disease caused by mutations in desmosomal genes, such as DSP and LMNA encoding the nuclear envelope protein lamin AC.21,22 The latter may also present with conduction abnormalities (atrioventricular node and/or left bundle branch disease) and clinical or subclinical skeletal myopathy. An increased risk of sudden cardiac death has also been identified in cases with mutations in desmosomal genes and LMNA, as compared to other genetic and non-genetic causes of DCM.23,24
Myocardial Inflammation
Ever since the first reports of AC, inflammatory cell infiltrates have been described in patients diagnosed at post-mortem, with the highest incidence in patients with more diffuse disease.3,25–29 Inflammatory infiltrates are also described in the transgenic models of AC.30,31 The subepicardial distribution of the typical AC lesions is very similar to that reported in myocarditis, and on occasion, patients with AC seek medical attention with symptoms of chest pain and palpitations accompanied by electrocardiographic changes and cardiac biomarkers consistent with myocarditis.32,33 These ‘hot phases’ can be the initial presentation of AC, sometimes preceding the development of classical ECG, imaging and histological features.34–37 18F-fluorodeoxyglucose PET can be positive in a subset of symptomatic AC patients, particularly in cases with left-dominant AC.32 Cardiac MRI in AC often demonstrates patterns of subepicardial scarring with a predilection for the inferior and lateral walls of the LV, similar to that seen following myocarditis.36–38 There are two studies showing raised cytokine levels in the blood of patients compared to controls.38,39
Cardiac sarcoidosis can show marked resemblance to genetic forms of AC, and the differential diagnosis is sometimes only made at a histological level.32 Furthermore, a link at the molecular level is also present, as both giant cell myocarditis and sarcoidosis exhibit a similar pattern of reduced plakoglobin staining in the absence of any mutations in the desmosomal genes.38 Finally, there is recent evidence of cardiac autoimmunity in a study of AC reporting anti-desmoglein antibodies associated with the extent of the disease.40 Importantly, this was a consistent finding among desmosomal and non-desmosomal forms of the disease.40
Approach to Diagnosis
Identifying phenotypes and defining the discrepancies between them might seem as an overly academic and mundane task; however, these concepts and their application are fundamental to treatment decisions and guide clinical and basic research.
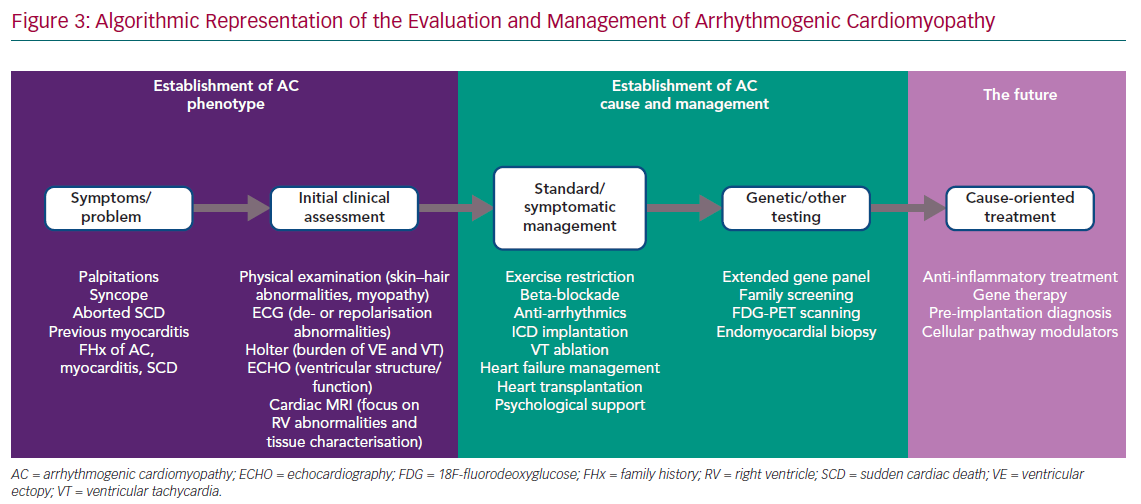
The phenotypic spectrum encompassed by ‘classic’ ARVC, biventricular AC and left-dominant AC challenges the development of a single set of diagnostic criteria. However, a stepwise approach to first identify the broad AC phenotype, as defined above, and then to characterise the genetic and non-genetic causes is possible in everyday practice, although the involvement of different specialities, such as clinical genetics, microbiology, immunology and haematology, is also essential in specific cases (Figure 3).
In this way, it is possible to move beyond the crude phenotypes currently used to define disease (e.g. heart failure with reduced or preserved ejection fraction) towards personalised algorithms for investigation and management.
Opportunities for Improved Management and New Therapies
The elucidation of specific subtypes of AC offers opportunities for tailored therapies. Examples where this is already possible include arrhythmia risk stratification, myocarditis treatment and the role of exercise.
Arrhythmia Risk Stratification
Risk stratification in AC is critical, as ICDs provide a life-saving therapy for patients prone to haemodynamically unstable ventricular arrhythmias.41 It is uncontentious that patients with previous unstable ventricular arrhythmias are at high risk of recurrence and an ICD is always recommended for secondary prevention.41 Although there have been numerous studies on arrhythmic risk stratification in affected individuals, and particularly family members from cascade screening with no previous arrhythmic events, the timing of ICD implantation remains a challenge.42–45 A new prediction model for ventricular arrhythmias (https://arvcrisk.com) has been recently developed and published by a multi-centre cohort. However, this is based only on individuals with a definite diagnosis of ARVC based on the 2010 Task Force criteria, and its utility remains obscure for the left-dominant forms.42
In a recent report, the Heart Rhythm Society provided consensus recommendations on the management of different forms of AC using a genotype-guided approach.18 Evidence of increased arrhythmic risk in cases with only mildly impaired LV systolic function exist for specific genes, such as PLN, LMNA and FLNC, and ICD implantation should be considered, irrespective of symptoms, in such cases.18 In the case of LMNA, additional parameters, such as male sex and non-sustained VT, can guide decision-making.
Myocarditis Treatment
As mentioned previously, myocardial inflammation is an important component of the AC spectrum.32,46 Identification of specific forms of inflammation, such as sarcoidosis, can be challenging, as a definite diagnosis can be reached only by endomyocardial biopsy, but is critical as it can dictate further management with corticosteroid therapy and long-term immunosuppression.47–49 The effects of anti-inflammatory therapy in gene-positive individuals presenting with a ‘hot phase’ remains to be investigated.
The increasing recognition of myocardial inflammation as a mechanism of disease progression in genetic forms of AC raises several possibilities in regards to therapeutic approaches.37 Although conventional treatment options for myocarditis, such intravenous immunoglobulin, corticosteroids, cyclosporine and azathioprine, can be considered, they remain largely non-selective in terms of the mechanisms and the sites of inflammation that they target.50,51 The elucidation of the cellular mechanisms in the various genetic forms of AC will be crucial in the identification of the potential causes of inflammatory induction that can be targeted therapeutically.
The Role of Exercise
It is common to elicit a history of high-intensity exercise among individuals affected by classic ARVC. Furthermore, patients with ARVC who were athletes have been found to have increased risk of sudden death compared with non-athletes.52 Desmosomal mutation carriers who participate in endurance and frequent exercise have a higher incidence of ventricular tachyarrhythmias and heart failure compared to those who are less athletic.53 A history of vigorous exercise seems to have an even greater effect in patients without an identifiable mutation, and patients seem to have greater benefit from exercise restriction.54,55 Further research is warranted to better understand the role of exercise in other subtypes of AC and non-desmosomal mutation carriers to prescribe exercise restriction more effectively.
Future Developments in Mechanism-based
Therapies
Recent studies have revealed a key role of the glycogen synthase kinase-3 beta (GSK3B) in the pathogenesis of AC.31 In the hearts of patients with AC, it has been shown that GSK3B translocates from the cytoplasm, where it is present in controls, to the intercalated disc.31 This observation was consistent among all cases with desmosomal gene mutations, including DSP, but less consistent in FLNC and PLN mutation carriers.56,57 The inhibition of GSK3B led to rescue of the phenotype when administered early in transgenic mice with mutant DSG2 or JUP.31
In disease caused by LMNA mutations, a selective p38 mitogen-activated protein kinase inhibitor, ARRY-797, has shown reversal of cardiac dysfunction in a transgenic murine model of LMNA. A Phase III clinical trial is now underway in patients with DCM caused by LMNA mutations (NCT03439514).
Examples of other molecular-targeted treatments include gene therapy enabling trans-splicing in a LMNA-related muscular dystrophy mouse model with partial rescue of the mutant phenotype.58 Antisense-mediated exon skipping can remove mutant exons with frameshift or nonsense variants in cardiomyopathy genes, and has been used to eliminate TTN truncating variants in patient-derived induced pluripotent stem cell-derived cardiomyocytes (iPSC-CM).59 Gene correction using transcription activator-like effector nucleases has successfully corrected PLN variants in iPSC-CM by establishing the biallelic expression of the normal PLN transcripts.60
Conclusion
In the era of ‘-omic’ technologies and precision medicine, there are increasing opportunities to redefine diseases and test new therapies. AC serves as a prime example of a heterogeneous family of disorders in which there are multiple genetic and non-genetic causes amenable to existing and developing therapies that promise to slow, reverse and perhaps prevent disease.