









Statins are most frequently prescribed for the primary prevention of cardiovascular disease due to their lipid-lowering properties. They are known to exert their effect by competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. The reduction in the synthesis of cholesterol induces upregulation of LDL receptors in the liver, resulting in increased clearance and decreased deposits of LDL in blood vessels.1 These LDL deposits are central to the induction of atherogenesis once oxidised by reactive oxygen species.2 Reducing circulating LDL leads to less oxidised LDL, which causes less damage.
However, it was discovered that the therapeutic effect of statins is not solely due to the reduction of LDL. This is because statins are able to reduce mortality in healthy individuals with a normal LDL cholesterol profile.3 Similarly, other cholesterol-lowering methods, such as ileal bypass surgery or bile acid sequestrants, do not have an immediate clinically beneficial result, despite the drop in LDL.4
Statins also show a significant reduction in morbidity, mortality, recurrent unstable angina incidents, non-fatal MI after 4 months and reduced rehospitalisation in just 30 days for acute coronary syndrome (ACS) patients.5–9 Studies also demonstrated that the use of a higher statin dose strategy did not increase the clinical benefit, despite greater LDL reduction.10
Apart from their lipid-lowering ability, statins have various immunomodulatory properties that have recently been identified.11 Inhibition of HMG-CoA reductase not only suppresses cholesterol synthesis, but also various inflammatory pathways due to the intermediates in the mevalonate pathway. Two isoprenoids, farnesyl-pyrophosphate and geranylgeranyl-pyrophosphate, are involved in post-translational modifications, in particular, the prenylation of GTPases, such as Ras, Rac, Rho and Cdc42.12 These binary switches are involved in various inflammatory pathways activated by their effect on cell signalling via phophatidylinositol-3-kinase, mitogen-activated protein kinase and nuclear factor kappa-light chain-enhancer of activated B-cells.
Of note, Rho activates nuclear factor kappa-light-chain-enhancer of activated B-cells, which reduces endothelial nitric oxide synthase.13 Inhibition by statins would increase nitric oxide bioavailability and improve vascular function, which could explain how statins are able to improve vascular endothelial function within 3 hours of treatment in healthy patients.14 Inhibition of these pathways also causes a downregulation of vascular cell adhesion protein 1 and tumour necrosis factor-alpha-induced expression of intercellular adhesion molecule 1, reducing the ability of macrophages to bind to endothelial cells.15 Macrophage activity is also disrupted because of the reduction in chemoattractants, such as monocyte chemoattractant protein 1.
Statins have also been shown to favour expression of anti-inflammatory cytokines, such as interleukin (IL)-4, IL-10 and transforming growth factor-beta (TGF-beta), while downregulating interferon-gamma (IFN-gamma), IL-6, IL-1 and IL-8 due to a shift from T-helper cell type 1 to T-helper cell type 2 response, as shown in a mouse model of autoimmune encephalomyelitis treated with statins.16 A similar milieu of anti-inflammatory mediators was seen to improve graft versus host disease in animal models with the treatment of atorvastatin due to its ability to inhibit protein geranylgeranylation.17
Independent of HMG-CoA reductase inhibition, statins have been shown to bind to an allosteric site on lymphocyte function-associated antigen 1, thereby blocking the recruitment of lymphocytes and their cytotoxicity.18 All the effects of statins highlighted demonstrate that statins tilt the balance towards an anti-inflammatory state, so it is not surprising when statins are shown to significantly reduce regulatory T-cells (Tregs) in healthy individuals.19
Tregs are a subpopulation of CD4+ T-cells that are induced by an inflammatory stimulus in the peripheries from naive CD4+ cells, but they can also be derived in the thymus as CD4+CD25− cells.20 It has been found that 5–10% of circulating CD4+ T-cells are Tregs.21 They are characterised by the constitutive expression of CD25 and CTLA4 receptors, and FOXP3; a transcription factor that promotes T-cell differentiation into Tregs.22,23 Although these markers may also be found on classic CD4+ T-cells,24,25 research for a specific marker of Tregs has identified a demethylation site in a 5’untranslated region of FOXP3 called Treg-specific demethylated region, which allows for discrimination of Tregs.26
Tregs are regarded as the master switch of immune system regulation by exerting their effect through the modulation of both adaptive and innate immune responses through multiple mechanisms. The main four mechanisms by which Tregs exert their effects are through the release of inhibitory cytokines, antigen-dependent inhibition of immune responses, and direct contact and inhibition of APC maturation, as well as the interaction with CD80/86 complex on APC via CTLA4.
Tregs release immunosuppressive cytokines IL-10, IL-35 and TGF-beta. Secretion of IL-10 by Tregs inhibits synthesis of pro-inflammatory cytokines (such as IL-6 and IL-2)and expression of major histocompatibility complex class II by APC, preventing antigen presentation.27 IL-35 secretion mediates induction of CD4+ cells into Tregs while also inhibiting proliferation of T-cells.28 TGF-beta released by Tregs decreases CD28 expression (a co-stimulatory molecule required for the activation of T-cells) and induces CD4+ differentiation into Tregs.29
Another functional mechanism of Tregs is antigen-dependent suppression. Tregs can be activated by an antigen via its T-cell receptor, where after it may suppress a T-cell with any antigen specificity.30 Direct contact of Tregs with APC exerts multiple actions. One of them is downregulation of co-stimulatory ligands CD80/CD86, which are required for T-cell activation.31 They also prevent dendritic cell activation by inhibiting effector T-helper cell type 1 cells (reducing IFN-gamma and tumour necrosis factor-alpha production), which is required for antigen presentation.32
Tregs also modulate macrophage activity, in particular, reducing their production of matrix metalloproteinases.33 However, the predominant mechanism by which Tregs exert their inhibitory effect on immune responses is through expression of CTLA4 molecule on its surface. CTLA4 binds the CD80/86 complex on APC, hence preventing CD28-mediated activation of CD4+T-helper cells, leading to downregulation of both B-cell and cytotoxic T-lymphocyte (CTL)-mediated inflammatory responses.34 Binding also generates inhibitory signals to decrease the expression of CD28 while enhancing FOXP3 expression.35
Finally, CTLA4 and CD80/CD86 binding can also induce expression of indoleamine 2,3-dioxygenase, which catabolises tryptophan into pro-apoptotic mediators.36 Other mechanisms include Treg-mediated cytolysis of B-cells, natural killer cells and CTL via the release of granzyme A and perforin.37 Tregs can also cause metabolic disruption of CTL, as high levels of CD25 on Tregs cause depletion of IL-2 in the local environment, starving the effector T-cells and inducing apoptosis.38 Overall, Tregs play a significant role in the prevention of autoimmunity and induction of peripheral tolerance, but overactivation may also prevent immunity to certain pathogens or tumours.
Vascular endothelial injury and dysfunction are the initial stages of coronary atheroma formation. The influx of LDL and its oxidisation within the coronary vessels are followed by activation and recruitment of monocyte-derived macrophages, leading to formation of lipid-laden foam cells in the vessel lumen and early plaque lesions.2 The damaged endothelium and macrophages release cytokines and growth factors including IFN-gamma, platelet-derived growth factor and TGF-beta, which signal for the recruitment of pro-inflammatory T-cells and proliferation of smooth muscle cells.39
The chronic vicious cycle of inflammation and proliferation of collagen-secreting smooth muscle cells gradually leads to the thickening of the lesion and formation of a fibrolipid cap covering a highly thrombogenic necrotic core.40 Such advanced coronary plaques can, however, become destabilised and rupture through multiple inflammatory processes predominantly mediated by T-cells.41
In this context, an aggressive and unusual subpopulation of CD4+ T-cells, CD4+CD28− cells, has been reported to significantly contribute to coronary plaque destabilisation.42 These T-cells are believed to be activated by an auto-antigen, namely, human heat shock protein 60, expressed within the plaque environment. Human heat shock protein 60 is a chaperone stress protein that is constitutively expressed in all cells, and its expression is significantly increased during inflammation.42 Upon antigen exposure and activation, these T-cells are able to directly target and lyse the smooth muscle cell constituent of the plaque through the release of cytotoxic granules, resulting in plaque rupture and exposure of its thrombogenic content to the coronary blood flow. Once activated, CD4+CD28− cells also produce IFN-gamma, which activates local macrophages, leading to increased secretion of matrix metalloproteinase enzymes.43,44 These enzymes can gradually degrade the fibrous plaque cap, and contribute to plaque destabilisation and rupture resulting in thrombosis and ACS.
ACS is characterised by heightened inflammatory status, and T-cells within the coronary environment, in particular, have been implicated in both disease initiation and progression.43,45 It has been shown that the frequency of Tregs is reduced in patients with non-ST-segment elevation MI, ST-segment elevation MI, acute MI and unstable angina.46,47 In addition, Wigren et al. showed that individuals with low levels of Tregs were at increased risk of a first coronary event.48 Furthermore, Tregs are reported to have compromised functional efficacy, increased tendency for apoptosis and reduced responsiveness to T-cell receptor-mediated induction.46,49,50 These findings have led to suggestions that Tregs are unable to modulate coronary disease progression through tilting the internal milieu in support of a pro-inflammatory state.
ACS patients have been shown to benefit from the anti-inflammatory effects of statin treatment; although, the exact mechanisms by which these effects are exerted are still debatable. Since ACS is characterised by diminished frequency and function of Tregs, their induction could potentially shift the immunomodulatory balance to an anti-inflammatory state. To date, there is no systematic review and meta-analysis compiling results from different randomised controlled trials (RCTs) in ACS patients.
The aim of this systematic review and meta-analysis is to evaluate whether statin therapy enhances the frequency of CD4+CD25+FOXP3+ Tregs in patients with ACS.
Methods
Eligibility Criteria
Studies were restricted to RCTs, with no minimum or maximum length of follow-up. Adults diagnosed with ACS were included in this study. Studies that quantified Tregs cell frequency by flow cytometric analysis before and after statin treatment were included. A minimum of at least two of the conventional markers from CD4+CD25+FOXP3+ was compulsory to identify Tregs.
The intervention must have been any of the oral tablet forms of statins: simvastatin, atorvastatin, rosuvastatin, pravastatin and fluvastatin, of which at least a minimum of 10 mg and a maximum of 80 mg dosage were prescribed
Search Strategy
Studies from all years and languages were allowed for inclusion. MeSH terms were used to add synonyms and to increase the scope of the search (Supplementary Material Appendix 1). Following identification of eligibility criteria and the relevant search terms, the PubMed (1996 to search date) and EMBASE (1947 to search date) databases were searched on 13 June 2017. The detailed search strategy can also be found in Supplementary Material Appendix 2.
Data Collection and Analysis
The title and abstract of all studies retrieved were evaluated for relevance to the objective of this review using the population, intervention, comparator, outcomes and study type (Supplementary Material Appendix 1). The piloted spread sheet contained the population, intervention, comparator, outcomes and study type, p-value, power, mean, Treg percentage, SD, 95% CI and length of the study.
Quality Assessment of Included Studies
To assess the risk bias of eligible RCTs, authors (NS and HA) used the Cochrane risk of bias tool. The randomisation, allocation concealment, blinding of patients, researchers, outcome, completeness of data reported and selection of biomarkers used to identify Treg cells were evaluated. Studies should state the patient characteristics, exclusion criteria, lost to follow-up and number of patients in each group. If studies did not use all three CD25, CD4 and FOXP3 biomarkers, they were classified as high risk of information bias, as this may increase the probability of an impure yield of Tregs. Studies needed to report that division of participants into control or statin therapy groups had been conducted randomly. However, a detailed account of randomisation was not required, as long as the patient characteristics of both groups were included and did not differ significantly (p>0.05).

Synthesis of Results
Standardised mean difference (SMD) was used as a summary measure due to the nature of the outcomes. As the data type is continuous, the statistical method to pool the results will be inverse variance.
Assessment of Homogeneity
Chi-squared and I-squared were calculated using Review Manager 5.3. Heterogeneity was interpreted according to guidelines by the Cochrane Handbook 5.1.0. A random effects model was used, as the I-squared was significant (>50%). Due to the significant heterogeneity, a subgroup analysis was performed by the type of statin. A meta-regression was also performed to see the dose-related effect.
Results
Study Selection
Search Results
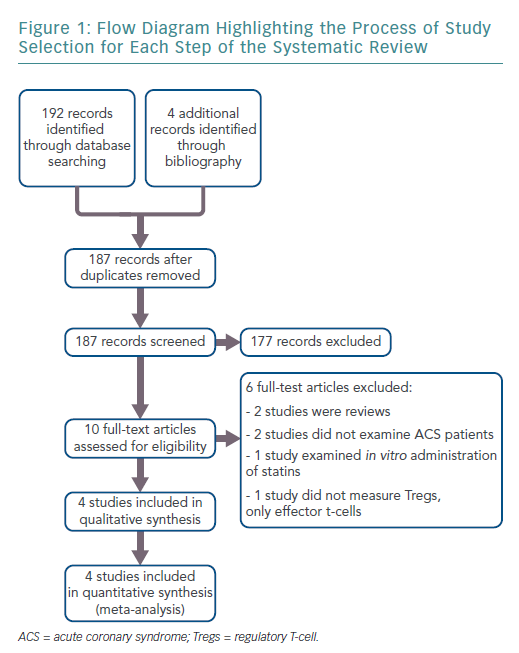
The search results retrieved 192 articles. Four additional records were identified through bibliographies. After duplicates were removed, 187 articles remained. Ten full-text articles were reviewed for inclusion, of which four met the full eligibility criteria. Figure 1 shows the flow diagram for study selection.
Characteristics of Included Studies
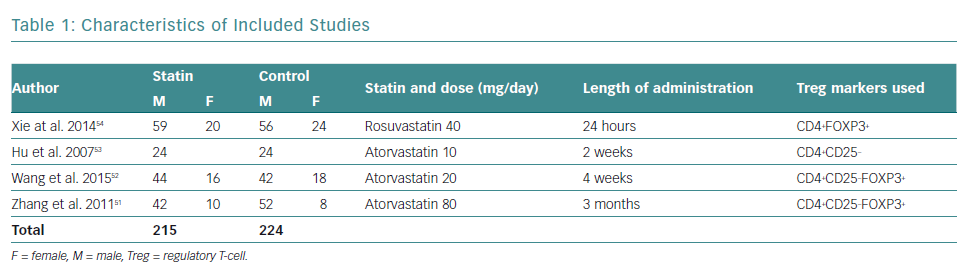
There was a total of 439 participants from all the included studies, of which 215 received statin therapy and 224 were part of the control groups. The characteristics of the included studies are summarised in Table 1.
All studies were RCTs reported in English. Three studies administered atorvastatin oral tablets, one at a dose of 80 mg, and the rest at 20 mg and 10 mg, respectively.51–53 A study by Xie et al. used rosuvastatin oral tablets at a dose of 40 mg.54 Only studies by Zhang et al. and Wang et al. used placebos in the control group, compared with Hu et al. and Xie et al., who did not administer any medication to the control group, besides conventional ACS medication.51–54
Risk of Bias
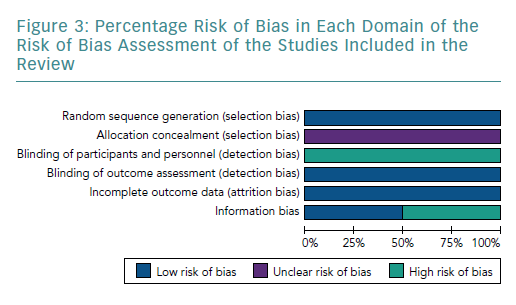
Random Sequence Generation
The four studies had a low risk of bias due to randomisation (Figures 2 and 3).51–54
Allocation Concealment
None of the studies provided adequate information to assign bias as low or high, consequentially the risk of bias remains unclear.51–54
Blinding
All studies had a high risk of bias by blinding due to the nature of the study, as blinding was not possible.51–54
Incomplete Outcome Data
All studies included had a low risk of incomplete outcome data, as all results were reported for outcomes identified at the start of the review.51–54
Information Bias
Two studies used the three known markers, CD4, CD25 and FOXP3, to identify Tregs.51,52 Therefore, they had a low risk of bias. In contrast, Hu et al. and Xie et al. only used two markers each – CD4 and CD25, and CD4 and FOXP3, respectively.53,54 Both Hu et al. and Xie et al. used fewer surface markers to identify Tregs, which increased the chance of an impure yield. Further details of the rationale of bias assessment are found in Supplementary Material Appendix 3.
Synthesis of Results
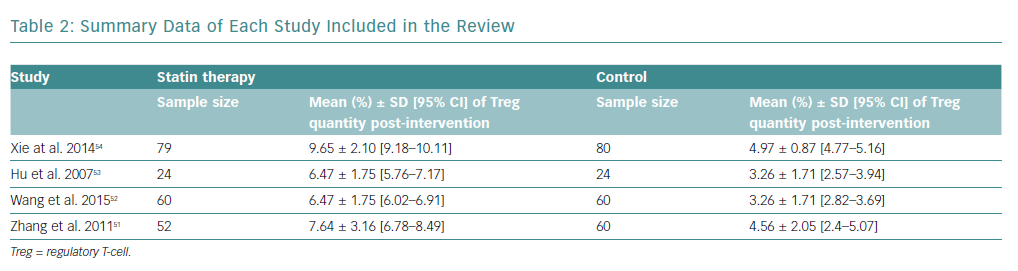
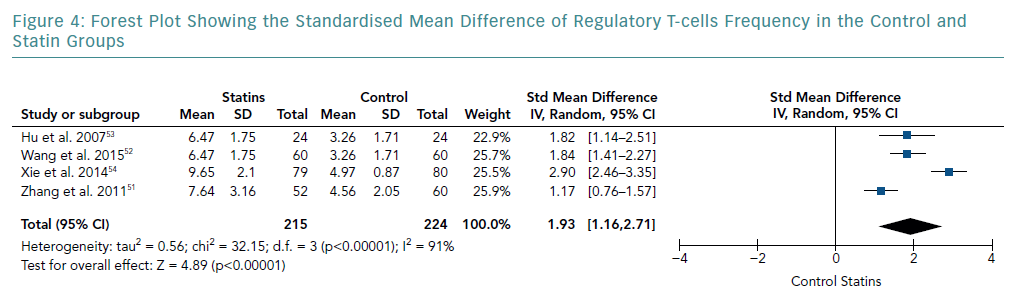
There was an increase in the mean frequency of Treg percentage in patients who received statins in all four studies (Table 2). Each study reported a p<0.01 for Treg frequency. Meta-analysis for Treg frequency demonstrated significantly higher values in patients treated with statins (SMD 1.93; 95% CI [1.16–2.71]; p<0.00001; Figure 4).
Heterogeneity of Results
The heterogeneity of effect sizes resulted in a I-squared value of 91%. These results signify that the effect sizes are heterogeneous with very high inconsistency in the data.
Subgroup Analysis
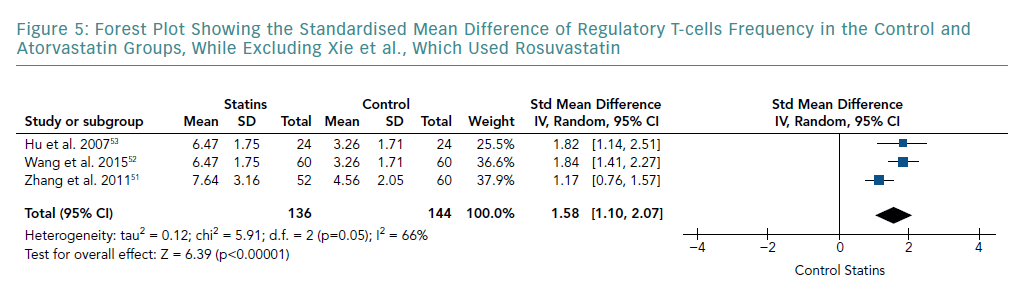
A subgroup analysis of studies that only used atorvastatin is shown in Figure 5. This was carried out by removing the study by Xie et al., which used rosuvastatin. The meta-analysis still indicated a significant increase in Treg frequency compared with the control group (SMD 1.58; 95% CI [1.10–2.07]; p<0.00001). However, pooled results showed a lower SMD than that of all the studies combined. Although this subgroup analysis showed a further decrease in heterogeneity (I-squared from 91% to 66%), it still remained significant.
Meta-regression
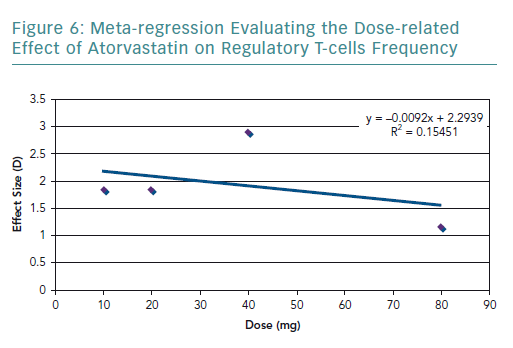
To further explore the high heterogeneity values, dosage-related effect was explored through a meta-regression (Figure 6). The meta-regression including all the studies resulted in an R2 value of 0.15 and correlation coefficient of −0.39. The graph showed a dose-related effect; however, there was an outlier – the study by Xie et al. – which used 40 mg of rosuvastatin.54
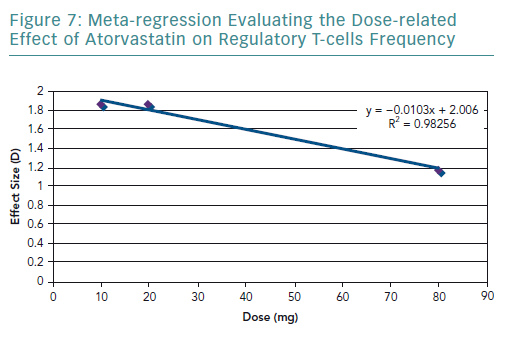
A meta-regression of the other three studies that used atorvastatin was conducted. It resulted in an R2 value of 0.98 (Figure 7) and correlation coefficient of −0.99. These values are very close to 1, suggesting a strong negative relationship.
Discussion
The results of the present review highlighted that statins significantly increase the frequency of Tregs. However, there is a considerable amount of heterogeneity in the pooled results, which was explored in terms of the statin type and dosage used. In terms of statin type, three of the studies used atorvastatin in the experimental group, and only Xie et al. used rosuvastatin.54 By removing Xie et al., the heterogeneity decreased, indicating that the effect on the frequency of Tregs varies according to the statin type. The dose-related effect was evaluated by conducting a meta-regression. It was found that there is a negative correlation with dose and the frequency of Tregs. However, there were very few studies that used the same statin type and dose, limiting the amount of data available for statistical aggregation.
The studies included in the review have a high risk of information. In terms of information bias, the studies by Hu et al. and Xie et al. have a high risk because these did not use all the Tregs markers used to identify Treg cells, which may alter the results.53,54 However, even the studies that used all three or at least two of the classic markers (CD4, FOXP3 and CD25) may also have a risk of bias. This is because conventional CD4+ T-cells express FOXP3 transiently when activated and express CD25 upon stimulation.55–57 ACS patients are known to have heightened CTL, which correlates with the extent of the disease. Therefore, using biomarkers that may detect CTL in such patients could further increase the impurity of the results. However, due to the lack of studies, subgroup analysis could not be performed to measure whether the effect of statins on Tregs correlated with the different patterns of Treg markers used. Future studies need to use a more accurate method in identifying Tregs to produce more reliable results. The use of the FOXP3 locus (Treg-specific demethylated region) is recommended as a specific marker of Tregs rather than the sole use of fluorescence-activated cell sorting.26
The present review is only applicable to the Asian population, as all studies were conducted in Asia. Moreover, it can only be applied to countries where the use of rosuvastatin and atorvastatin is approved.
Despite the limitations highlighted by the review, the potential of statins in inducing the frequency of Tregs has been shown by different studies with a plausible mechanistic effect. A similar benefit induced by statins was shown in rheumatoid arthritis, which is also characterised by impaired Tregs functioning.58 A RCT showed that atorvastatin treatment increases the amount of Tregs in the PBMC of rheumatoid arthritis patients and reduces disease activity.59
This trend was also seen in people with asthma, for whom statin therapy improved symptom control, and in vitro studies with the incubation of statins and CD4+ T-cells revealing expansion of Tregs.60,61 The effect of statins in upregulating of Tregs and reducing disease burden was also seen in animal models of autoimmune neuritis, experimental autoimmune myasthenia gravis (Li et al.), ischaemia-reperfusion injury and apolipoprotein E (ApoE)−/− model of atherosclerosis.62–65
However, Hasib et al. demonstrated that patients with ACS had a low baseline frequency of Tregs, and despite 12 months of extensive statin use after an acute event, Treg frequency remained significantly unchanged, which contradicts our findings.66 Although that was an observational study, it did not provide any strong support to the theory that statins exert a pronounced effect on Treg levels in a clinical scenario.
Finally, in terms of mechanism, Maussner-Fainberg et al. highlighted the ability of statins to induce FOXP3+ expression from peripheral CD4+CD25−FOXP3−.67 This expression may be induced by inhibition of HMG-CoA reductase, which suppresses intermediates in the mevalonate pathway leading to prenylation of GTPases, such as Ras, Rac, Rho and Cdc42.11–13 These small G proteins are postulated to be the reason for the induction of foxp3+ Tregs.67
Conclusion
ACS patients have been shown to clinically benefit from the anti-inflammatory effects of statin therapy through different pharmacological mechanisms. The current literature indicates a significant difference in Treg frequency following statin therapy in ACS patients. It is hypothesised that this is due to the stabilisation of the plaque by decreasing pro-inflammatory mediators involved in plaque destabilisation. It is proposed that future RCTs use accurate methods to identify Tregs and study the dose-related effects.
Overall, these results have considerable implications for patients who would benefit from restoring the Treg-induced immunomodulatory balance, with a view to exploring new therapeutic approaches to the management of disease progression in ACS.
Despite the limited data available and heterogeneity, the present review highlights areas for improvement, such as using more specific universal markers for Tregs, such as the Treg-specific demethylated region, and determining the statin type and dose-related effect.